

Hippocampal mGluR1-dependent long-term potentiation requires NAADP-mediated acidic store Ca2+ signaling
- PMID: 30482851
- PMCID: PMC6679716
- DOI: 10.1126/scisignal.aat9093
Hippocampal mGluR1-dependent long-term potentiation requires NAADP-mediated acidic store Ca2+ signaling
Abstract
Acidic organelles, such as endosomes and lysosomes, store Ca2+ that is released in response to intracellular increases in the second messenger nicotinic acid adenine dinucleotide phosphate (NAADP). In neurons, NAADP and Ca2+ signaling contribute to synaptic plasticity, a process of activity-dependent long-term potentiation (LTP) [or, alternatively, long-term depression (LTD)] of synaptic strength and neuronal transmission that is critical for neuronal function and memory formation. We explored the function of and mechanisms regulating acidic Ca2+ store signaling in murine hippocampal neurons. We found that metabotropic glutamate receptor 1 (mGluR1) was coupled to NAADP signaling that elicited Ca2+ release from acidic stores. In turn, this released Ca2+-mediated mGluR1-dependent LTP by transiently inhibiting SK-type K+ channels, possibly through the activation of protein phosphatase 2A. Genetically removing two-pore channels (TPCs), which are endolysosomal-specific ion channels, switched the polarity of plasticity from LTP to LTD, indicating the importance of specific receptor store coupling and providing mechanistic insight into how mGluR1 can produce both synaptic potentiation and synaptic depression.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
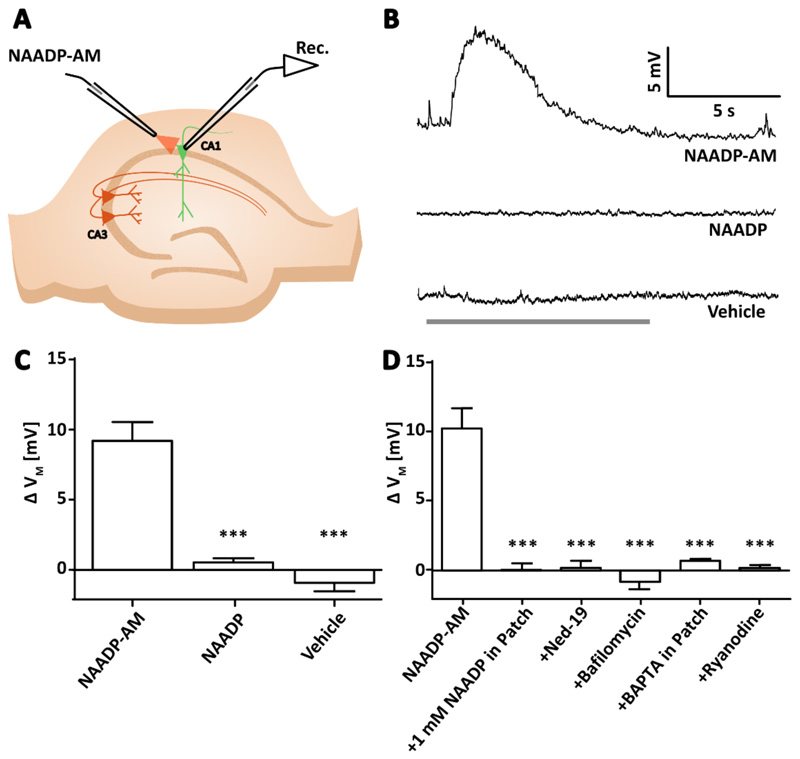
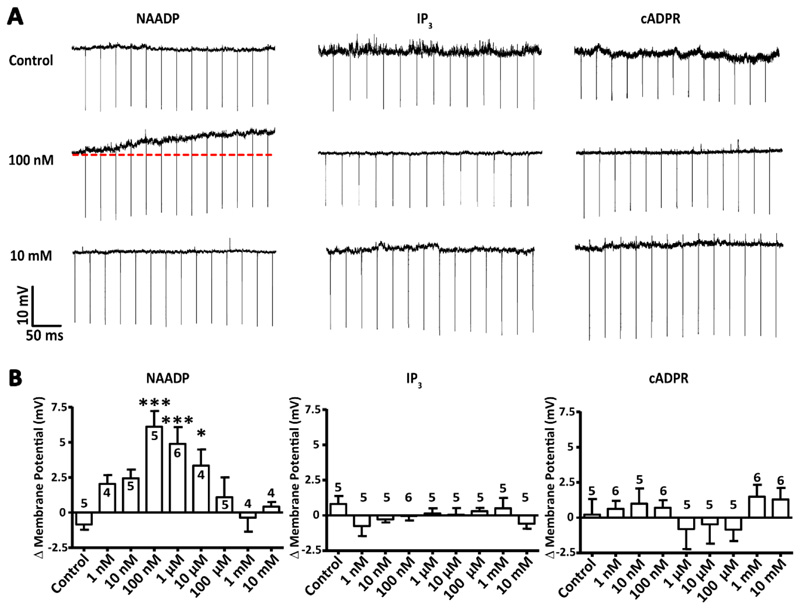
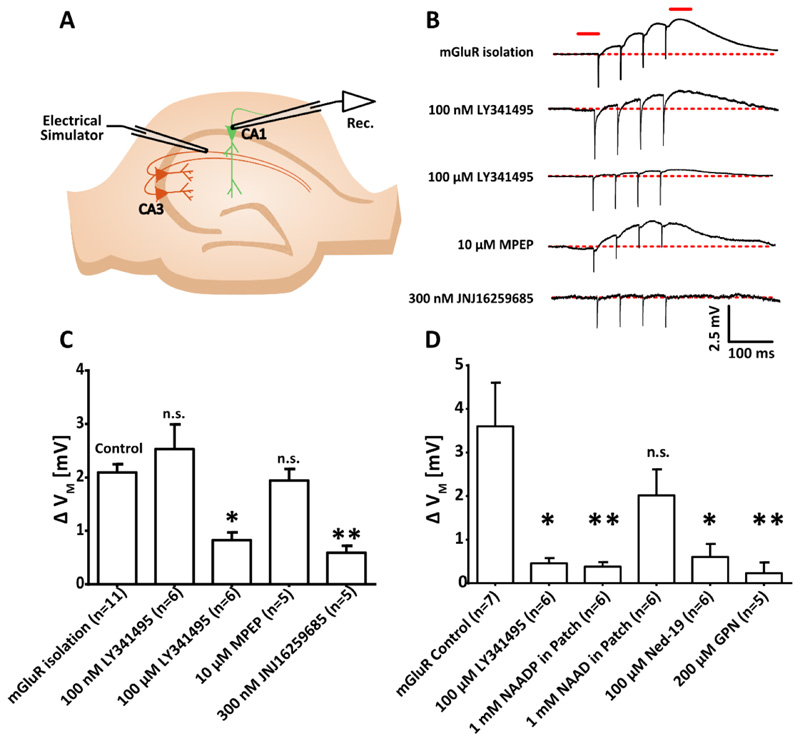
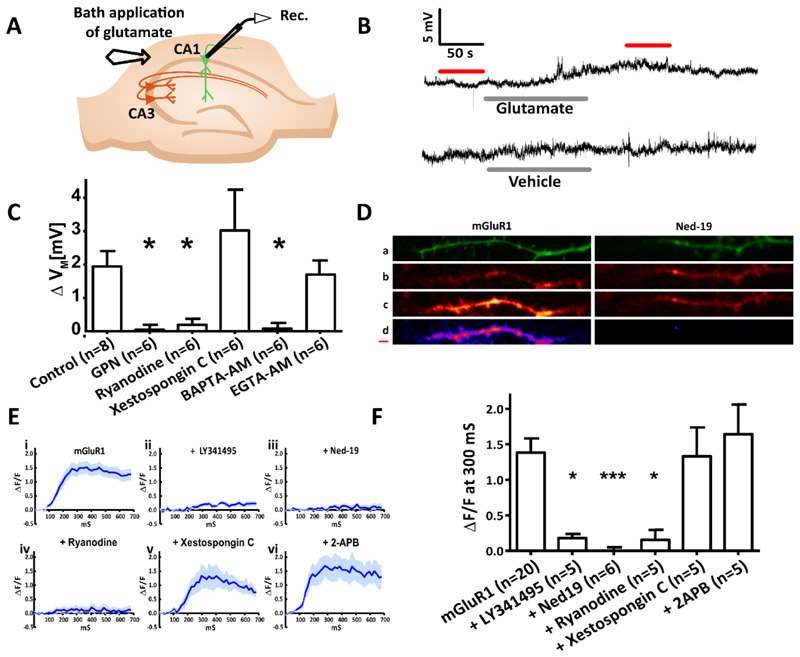
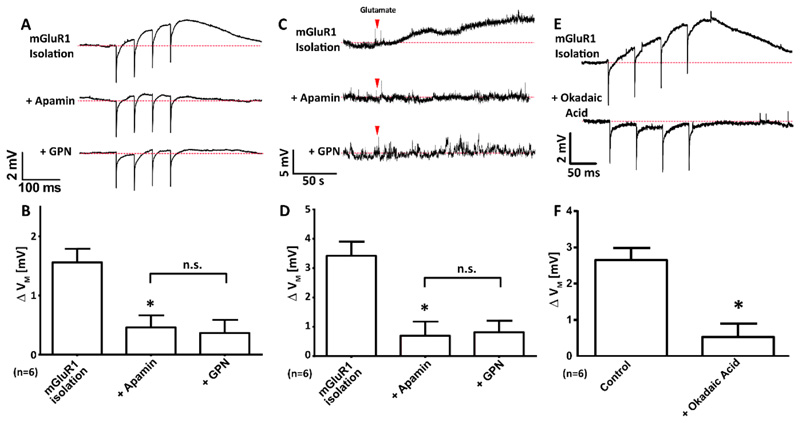
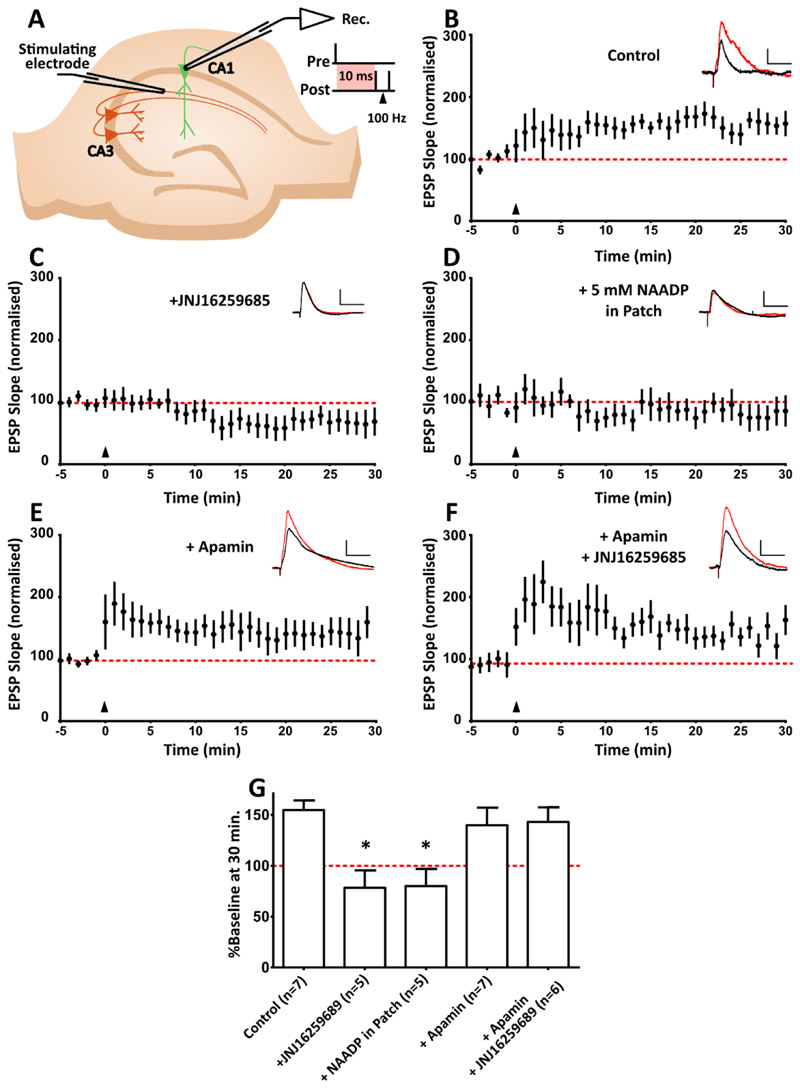
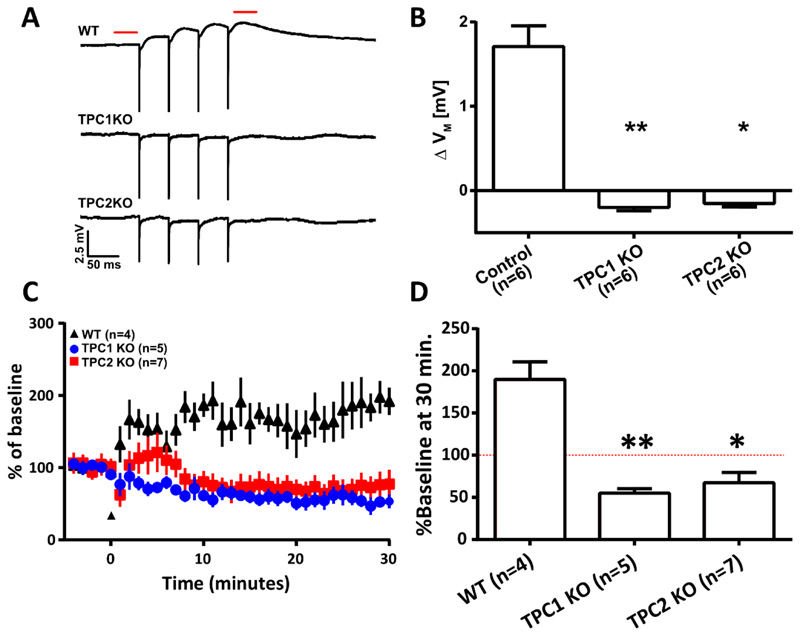
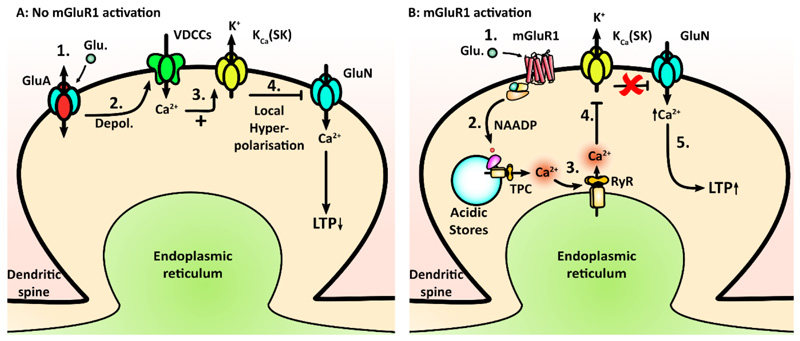
Comment in
-
Thinking through acidic Ca2+ stores.Sci Signal. 2018 Nov 27;11(558):eaau3342. doi: 10.1126/scisignal.aau3342. Sci Signal. 2018. PMID: 30482848
Similar articles
-
Both RyRs and TPCs are required for NAADP-induced intracellular Ca²⁺ release.Cell Calcium. 2015 Sep;58(3):237-45. doi: 10.1016/j.ceca.2015.05.005. Epub 2015 Jun 10. Cell Calcium. 2015. PMID: 26100948 Free PMC article.
-
Thinking through acidic Ca2+ stores.Sci Signal. 2018 Nov 27;11(558):eaau3342. doi: 10.1126/scisignal.aau3342. Sci Signal. 2018. PMID: 30482848
-
NAADP as an intracellular messenger regulating lysosomal calcium-release channels.Biochem Soc Trans. 2010 Dec;38(6):1424-31. doi: 10.1042/BST0381424. Biochem Soc Trans. 2010. PMID: 21118101
-
The acid test: the discovery of two-pore channels (TPCs) as NAADP-gated endolysosomal Ca(2+) release channels.Pflugers Arch. 2009 Sep;458(5):869-76. doi: 10.1007/s00424-009-0682-y. Epub 2009 May 28. Pflugers Arch. 2009. PMID: 19475418 Free PMC article. Review.
-
Modulation of Calcium Entry by the Endo-lysosomal System.Adv Exp Med Biol. 2016;898:423-47. doi: 10.1007/978-3-319-26974-0_18. Adv Exp Med Biol. 2016. PMID: 27161239 Review.
Cited by
-
Contribution of NAADP to Glutamate-Evoked Changes in Ca2+ Homeostasis in Mouse Hippocampal Neurons.Front Cell Dev Biol. 2020 Jun 25;8:496. doi: 10.3389/fcell.2020.00496. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32676502 Free PMC article.
-
OCaR1 endows exocytic vesicles with autoregulatory competence by preventing uncontrolled Ca2+ release, exocytosis, and pancreatic tissue damage.J Clin Invest. 2024 Apr 1;134(7):e169428. doi: 10.1172/JCI169428. J Clin Invest. 2024. PMID: 38557489 Free PMC article.
-
Genetic Inactivation of Two-Pore Channel 1 Impairs Spatial Learning and Memory.Behav Genet. 2020 Nov;50(6):401-410. doi: 10.1007/s10519-020-10011-1. Epub 2020 Sep 5. Behav Genet. 2020. PMID: 32889694 Free PMC article.
-
Effects of Blocking Multiple Sources of Calcium in Hippocampus During Spatial Learning and Memory Using a Rapid Acquisition Variant of the Morris Water Task.Hippocampus. 2025 Mar;35(2):e70006. doi: 10.1002/hipo.70006. Hippocampus. 2025. PMID: 40071661 Free PMC article.
-
Lysosomal TPC2 channels disrupt Ca2+ entry and dopaminergic function in models of LRRK2-Parkinson's disease.J Cell Biol. 2025 Jun 2;224(6):e202412055. doi: 10.1083/jcb.202412055. Epub 2025 Apr 25. J Cell Biol. 2025. PMID: 40279672 Free PMC article.
References
-
- Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J. 2011;439:349–374. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
