

Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults
- PMID: 30482864
- PMCID: PMC6310811
- DOI: 10.1073/pnas.1809700115
Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults
Abstract
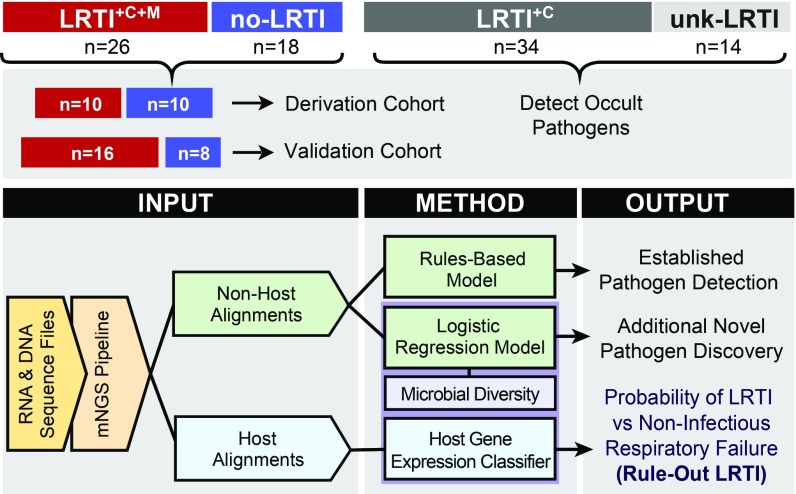
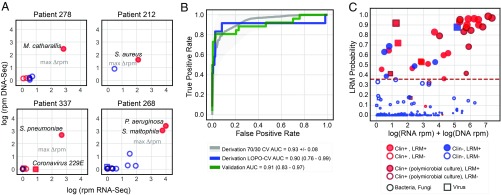
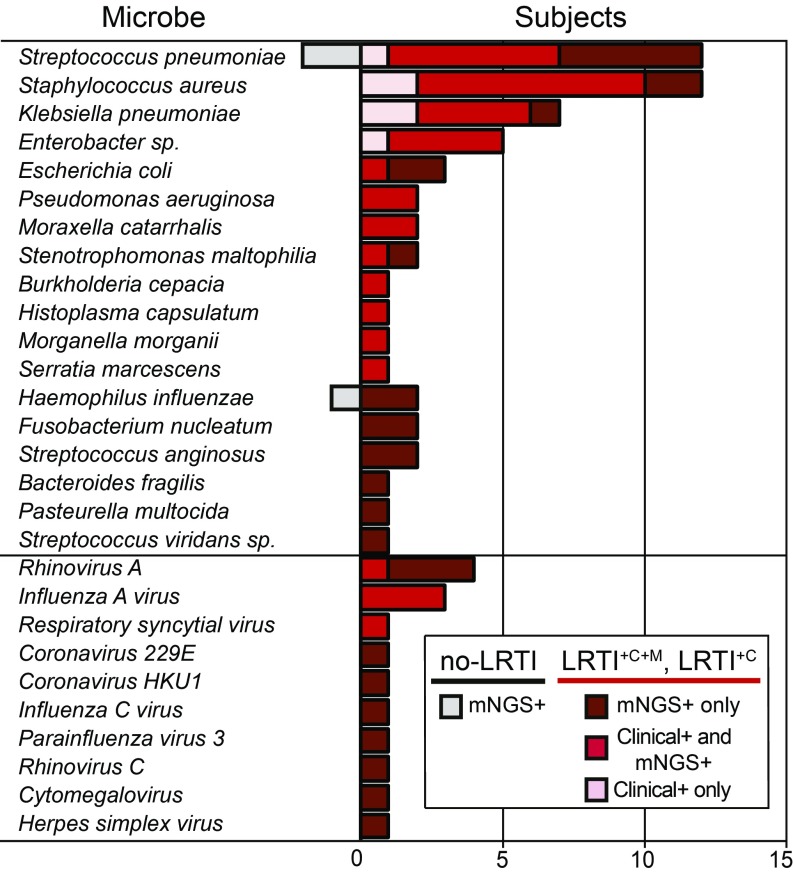
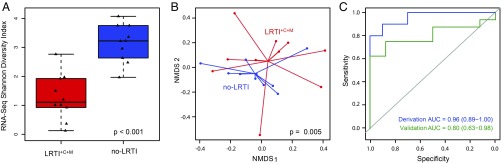
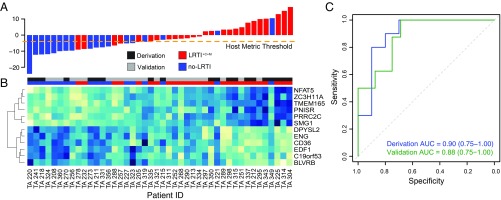
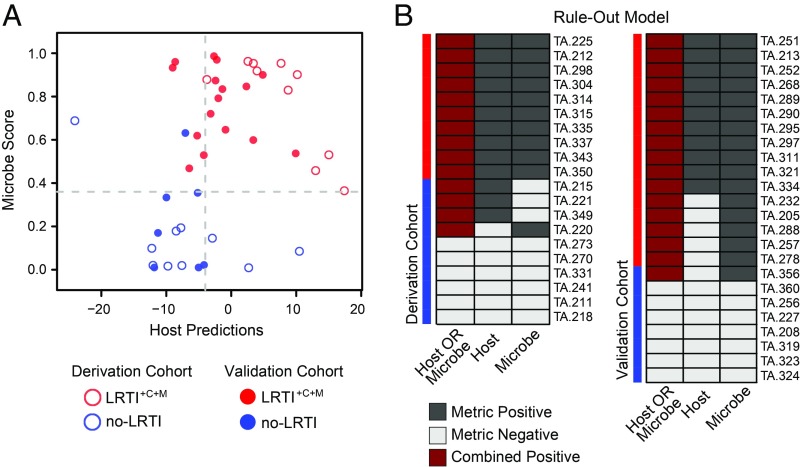
Lower respiratory tract infections (LRTIs) lead to more deaths each year than any other infectious disease category. Despite this, etiologic LRTI pathogens are infrequently identified due to limitations of existing microbiologic tests. In critically ill patients, noninfectious inflammatory syndromes resembling LRTIs further complicate diagnosis. To address the need for improved LRTI diagnostics, we performed metagenomic next-generation sequencing (mNGS) on tracheal aspirates from 92 adults with acute respiratory failure and simultaneously assessed pathogens, the airway microbiome, and the host transcriptome. To differentiate pathogens from respiratory commensals, we developed a rules-based model (RBM) and logistic regression model (LRM) in a derivation cohort of 20 patients with LRTIs or noninfectious acute respiratory illnesses. When tested in an independent validation cohort of 24 patients, both models achieved accuracies of 95.5%. We next developed pathogen, microbiome diversity, and host gene expression metrics to identify LRTI-positive patients and differentiate them from critically ill controls with noninfectious acute respiratory illnesses. When tested in the validation cohort, the pathogen metric performed with an area under the receiver-operating curve (AUC) of 0.96 (95% CI, 0.86-1.00), the diversity metric with an AUC of 0.80 (95% CI, 0.63-0.98), and the host transcriptional classifier with an AUC of 0.88 (95% CI, 0.75-1.00). Combining these achieved a negative predictive value of 100%. This study suggests that a single streamlined protocol offering an integrated genomic portrait of pathogen, microbiome, and host transcriptome may hold promise as a tool for LRTI diagnosis.
Keywords: lower respiratory tract infection; mechanical ventilation; next-generation sequencing; pneumonia; transcriptome.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Rethinking pneumonia: A paradigm shift with practical utility.Proc Natl Acad Sci U S A. 2018 Dec 26;115(52):13148-13150. doi: 10.1073/pnas.1819024116. Epub 2018 Dec 7. Proc Natl Acad Sci U S A. 2018. PMID: 30530647 Free PMC article. No abstract available.
References
-
- World Health Organization 2017 The top 10 causes of death. Available at www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death. Accessed October, 1, 2018.
-
- US Centers for Disease Control and Prevention 2018 Deaths: Leading Causes for 2016. Available at https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm. Accessed October 1, 2018.
-
- Zaas AK, et al. The current epidemiology and clinical decisions surrounding acute respiratory infections. Trends Mol Med. 2014;20:579–588. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
