

Human Calmodulin Mutations
- PMID: 30483049
- PMCID: PMC6243062
- DOI: 10.3389/fnmol.2018.00396
Human Calmodulin Mutations
Abstract
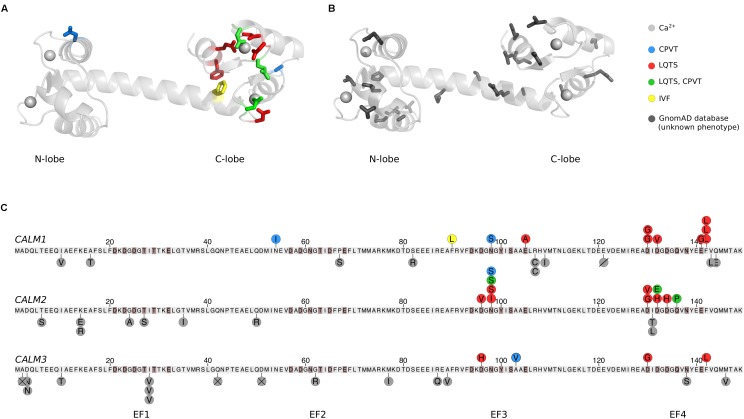
Fluxes of calcium (Ca2+) across cell membranes enable fast cellular responses. Calmodulin (CaM) senses local changes in Ca2+ concentration and relays the information to numerous interaction partners. The critical role of accurate Ca2+ signaling on cellular function is underscored by the fact that there are three independent CaM genes (CALM1-3) in the human genome. All three genes are functional and encode the exact same CaM protein. Moreover, CaM has a completely conserved amino acid sequence across all vertebrates. Given this degree of conservation, it was long thought that mutations in CaM were incompatible with life. It was therefore a big surprise when the first CaM mutations in humans were identified six years ago. Today, more than a dozen human CaM missense mutations have been described, all found in patients with severe cardiac arrhythmias. Biochemical studies have demonstrated differential effects on Ca2+ binding affinities for these CaM variants. Moreover, CaM regulation of central cardiac ion channels is impaired, including the voltage-gated Ca2+ channel, CaV1.2, and the sarcoplasmic reticulum Ca2+ release channel, ryanodine receptor isoform 2, RyR2. Currently, no non-cardiac phenotypes have been described for CaM variant carriers. However, sequencing of large human cohorts reveals a cumulative frequency of additional rare CaM mutations that raise the possibility of CaM variants not exclusively causing severe cardiac arrhythmias. Here, we provide an overview of the identified CaM variants and their known consequences for target regulation and cardiac disease phenotype. We discuss experimental data, patient genotypes and phenotypes as well as which questions remain open to understand this complexity.
Keywords: CALM1; CALM2; CALM3; CPVT; LQTS; calmodulin; calmodulinopathy; cardiac arrhythmia.
Figures
Similar articles
-
Spectrum and Prevalence of CALM1-, CALM2-, and CALM3-Encoded Calmodulin Variants in Long QT Syndrome and Functional Characterization of a Novel Long QT Syndrome-Associated Calmodulin Missense Variant, E141G.Circ Cardiovasc Genet. 2016 Apr;9(2):136-146. doi: 10.1161/CIRCGENETICS.115.001323. Epub 2016 Mar 11. Circ Cardiovasc Genet. 2016. PMID: 26969752 Free PMC article.
-
Calmodulin Mutations in Human Disease.Channels (Austin). 2023 Dec;17(1):2165278. doi: 10.1080/19336950.2023.2165278. Channels (Austin). 2023. PMID: 36629534 Free PMC article. Review.
-
Novel CALM3 Variant Causing Calmodulinopathy With Variable Expressivity in a 4-Generation Family.Circ Arrhythm Electrophysiol. 2022 Mar;15(3):e010572. doi: 10.1161/CIRCEP.121.010572. Epub 2022 Feb 28. Circ Arrhythm Electrophysiol. 2022. PMID: 35225649
-
Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks.Circ Arrhythm Electrophysiol. 2016 Aug;9(8):10.1161/CIRCEP.116.004161 e004161. doi: 10.1161/CIRCEP.116.004161. Circ Arrhythm Electrophysiol. 2016. PMID: 27516456 Free PMC article.
-
The emerging role of calmodulin regulation of RyR2 in controlling heart rhythm, the progression of heart failure and the antiarrhythmic action of dantrolene.Clin Exp Pharmacol Physiol. 2017 Jan;44(1):135-142. doi: 10.1111/1440-1681.12669. Clin Exp Pharmacol Physiol. 2017. PMID: 27626620 Review.
Cited by
-
Arrhythmia-associated calmodulin variants interact with KCNQ1 to confer aberrant membrane trafficking and function.PNAS Nexus. 2023 Oct 14;2(11):pgad335. doi: 10.1093/pnasnexus/pgad335. eCollection 2023 Nov. PNAS Nexus. 2023. PMID: 37965565 Free PMC article.
-
Understanding Calmodulin Variants Affecting Calcium-Dependent Inactivation of L-Type Calcium Channels through Whole-Cell Simulation of the Cardiac Ventricular Myocyte.Biomolecules. 2022 Dec 29;13(1):72. doi: 10.3390/biom13010072. Biomolecules. 2022. PMID: 36671457 Free PMC article.
-
Different arrhythmia-associated calmodulin mutations have distinct effects on cardiac SK channel regulation.J Gen Physiol. 2020 Dec 7;152(12):e202012667. doi: 10.1085/jgp.202012667. J Gen Physiol. 2020. PMID: 33211795 Free PMC article.
-
The mechanism of LQTS related CaM mutation E141G interfering with CaV1.2 channels function through its C-lobe.J Physiol Biochem. 2025 Feb;81(1):185-197. doi: 10.1007/s13105-024-01064-5. Epub 2024 Dec 19. J Physiol Biochem. 2025. PMID: 39699847
-
Impact of arrhythmogenic calmodulin variants on small conductance Ca2+ -activated K+ (SK3) channels.Physiol Rep. 2019 Oct;7(19):e14210. doi: 10.14814/phy2.14210. Physiol Rep. 2019. PMID: 31587513 Free PMC article.
References
-
- Berchtold M. W., Zacharias T., Kulej K., Wang K., Torggler R., Jespersen T., et al. (2016). The arrhythmogenic calmodulin mutation D129G dysregulates cell growth, calmodulin-dependent kinase II activity, and cardiac function in zebrafish. J. Biol. Chem. 291 26636–26646. 10.1074/jbc.M116.758680 - DOI - PMC - PubMed
-
- Boczek N. J., Gomez-Hurtado N., Ye D., Calvert M. L., Tester D. J., Kryshtal D. O., et al. (2016). Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ. Cardiovasc. Genet. 9 136–146. 10.1161/CIRCGENETICS.115.001323 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous