

Artificial Intelligence Understands Peptide Observability and Assists With Absolute Protein Quantification
- PMID: 30483279
- PMCID: PMC6242780
- DOI: 10.3389/fpls.2018.01559
Artificial Intelligence Understands Peptide Observability and Assists With Absolute Protein Quantification
Abstract
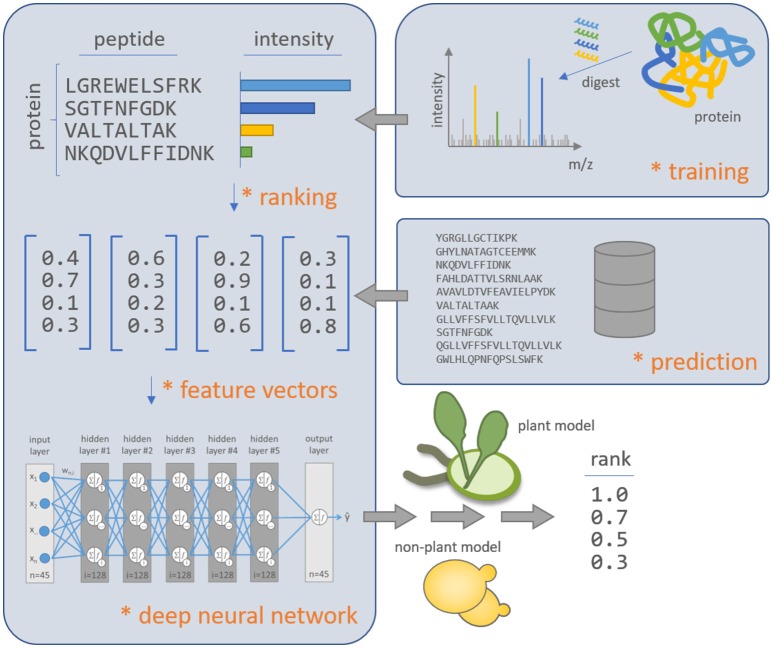
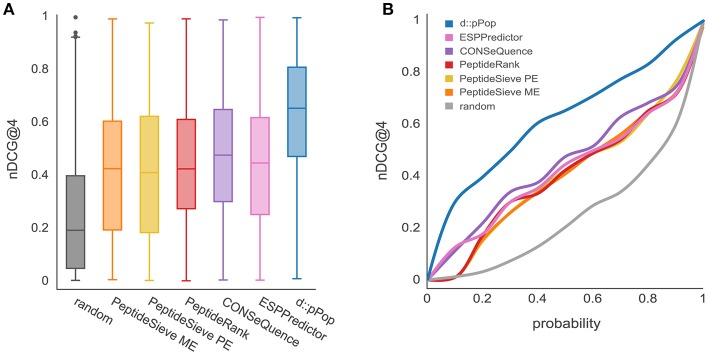
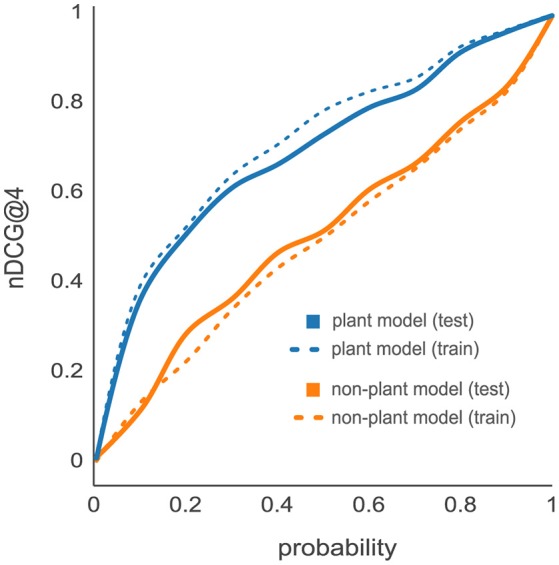
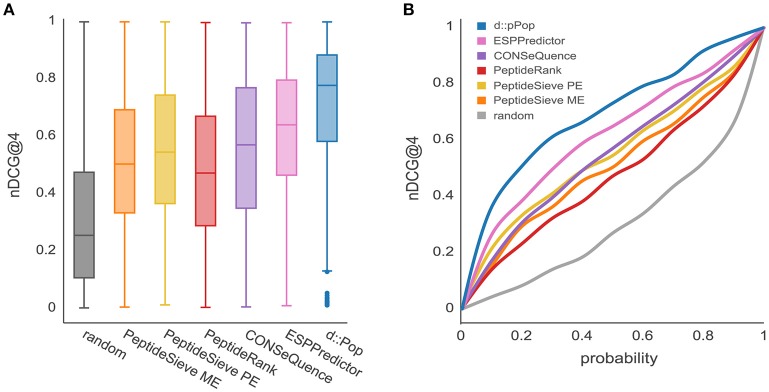
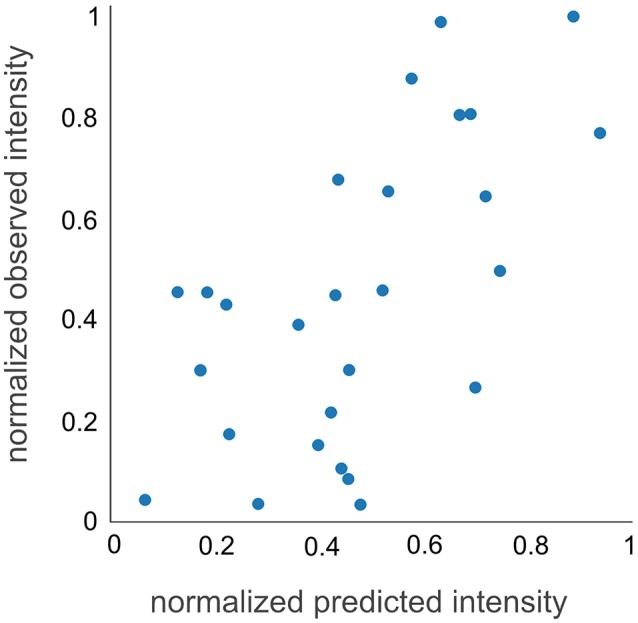
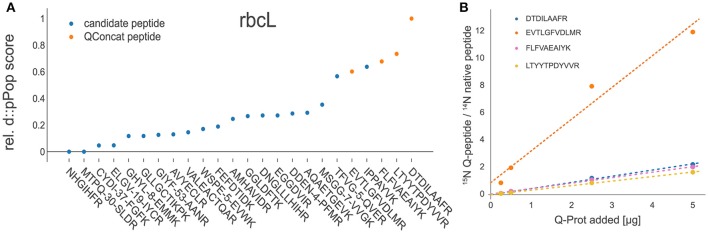
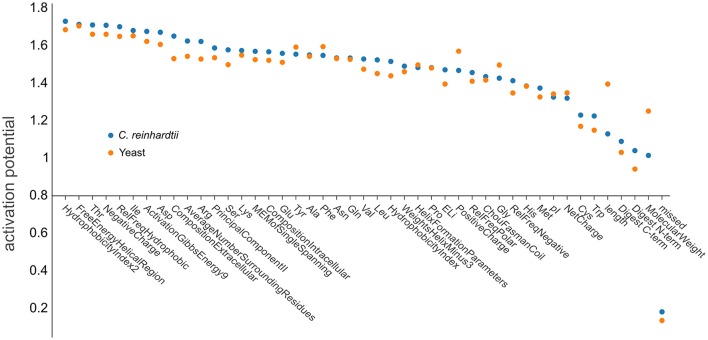
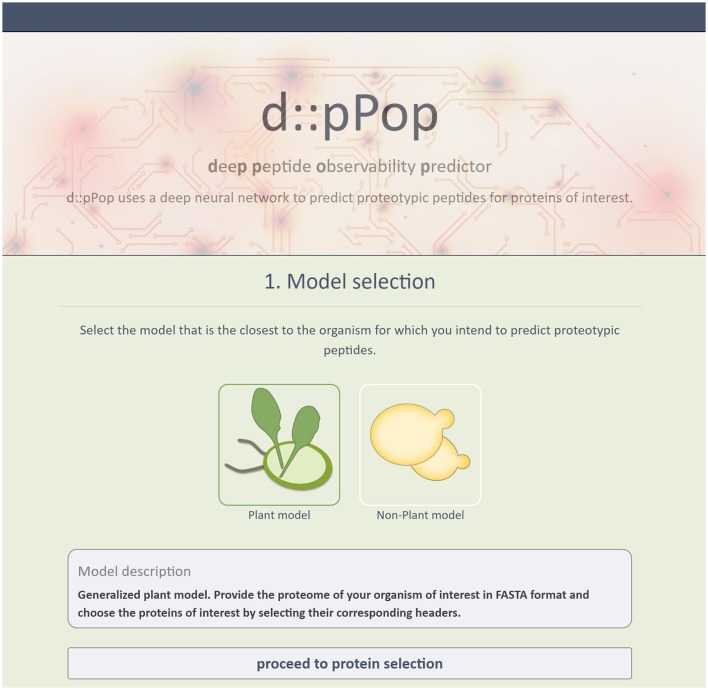
Targeted mass spectrometry has become the method of choice to gain absolute quantification information of high quality, which is essential for a quantitative understanding of biological systems. However, the design of absolute protein quantification assays remains challenging due to variations in peptide observability and incomplete knowledge about factors influencing peptide detectability. Here, we present a deep learning algorithm for peptide detectability prediction, d::pPop, which allows the informed selection of synthetic proteotypic peptides for the successful design of targeted proteomics quantification assays. The deep neural network is able to learn a regression model that relates the physicochemical properties of a peptide to its ion intensity detected by mass spectrometry. The approach makes use of experimentally detected deviations from the assumed equimolar abundance of all peptides derived from a given protein. Trained on extensive proteomics datasets, d::pPop's plant and non-plant specific models can predict the quality of proteotypic peptides for not yet experimentally identified proteins. Interrogating the deep neural network after learning from ~76,000 peptides per model organism allows to investigate the impact of different physicochemical properties on the observability of a peptide, thus providing insights into peptide observability as a multifaceted process. Empirical evaluation with rank accuracy metrics showed that our prediction approach outperforms existing algorithms. We circumvent the delicate step of selecting positive and negative training sets and at the same time also more closely reflect the need for selecting the top most promising peptides for targeting a protein of interest. Further, we used an artificial QconCAT protein to experimentally validate the observability prediction. Our proteotypic peptide prediction approach not only facilitates the design of absolute protein quantification assays via a user-friendly web interface but also enables the selection of proteotypic peptides for not yet observed proteins, hence rendering the tool especially useful for plant research.
Keywords: absolute quantification; deep learning; machine learning; mass spectrometry; peptide observability; proteotypic peptide.
Figures
References
-
- Barnidge D. R., Dratz E. A., Martin T., Bonilla L. E., Moran L. B., Lindall A. (2003). Absolute quantification of the G protein-coupled receptor rhodopsin by LC/MS/MS using proteolysis product peptides and synthetic peptide standards. Anal. Chem. 75, 445–451. - PubMed
LinkOut - more resources
Full Text Sources