

Using X-ray Footprinting and Mass Spectrometry to Study the Structure and Function of Membrane Proteins
- PMID: 30484402
- PMCID: PMC7780372
- DOI: 10.2174/0929866526666181128142401
Using X-ray Footprinting and Mass Spectrometry to Study the Structure and Function of Membrane Proteins
Abstract
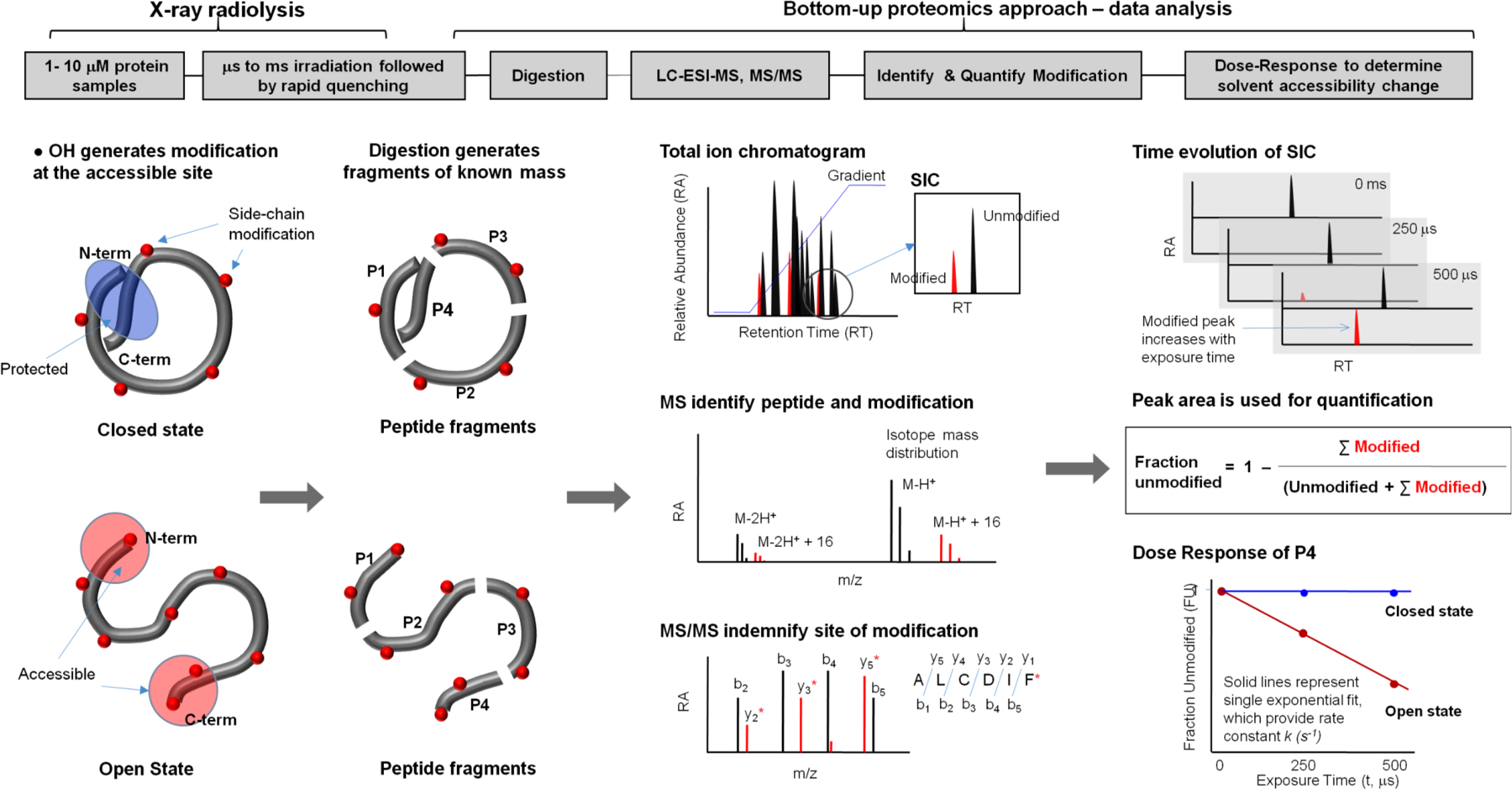
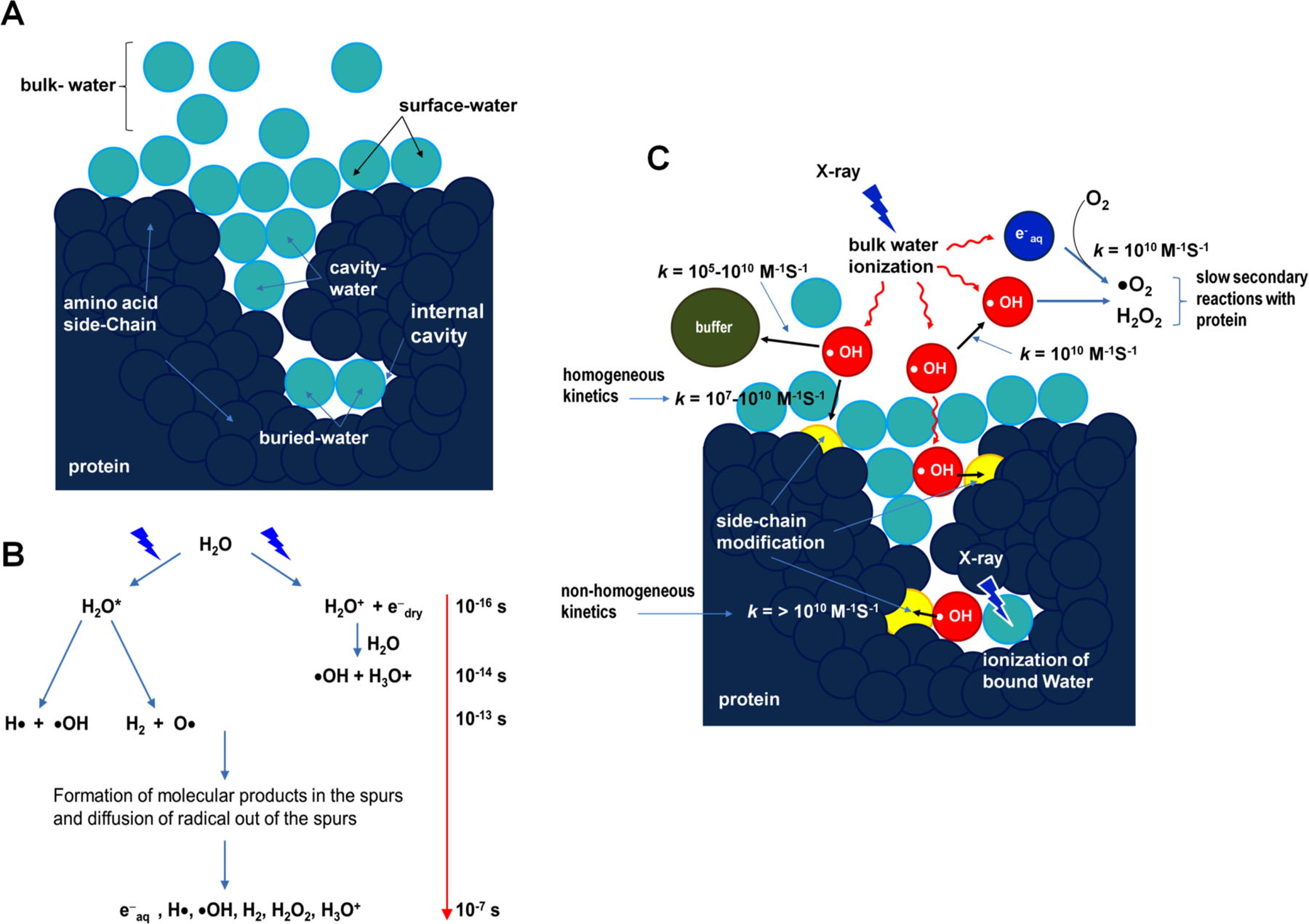
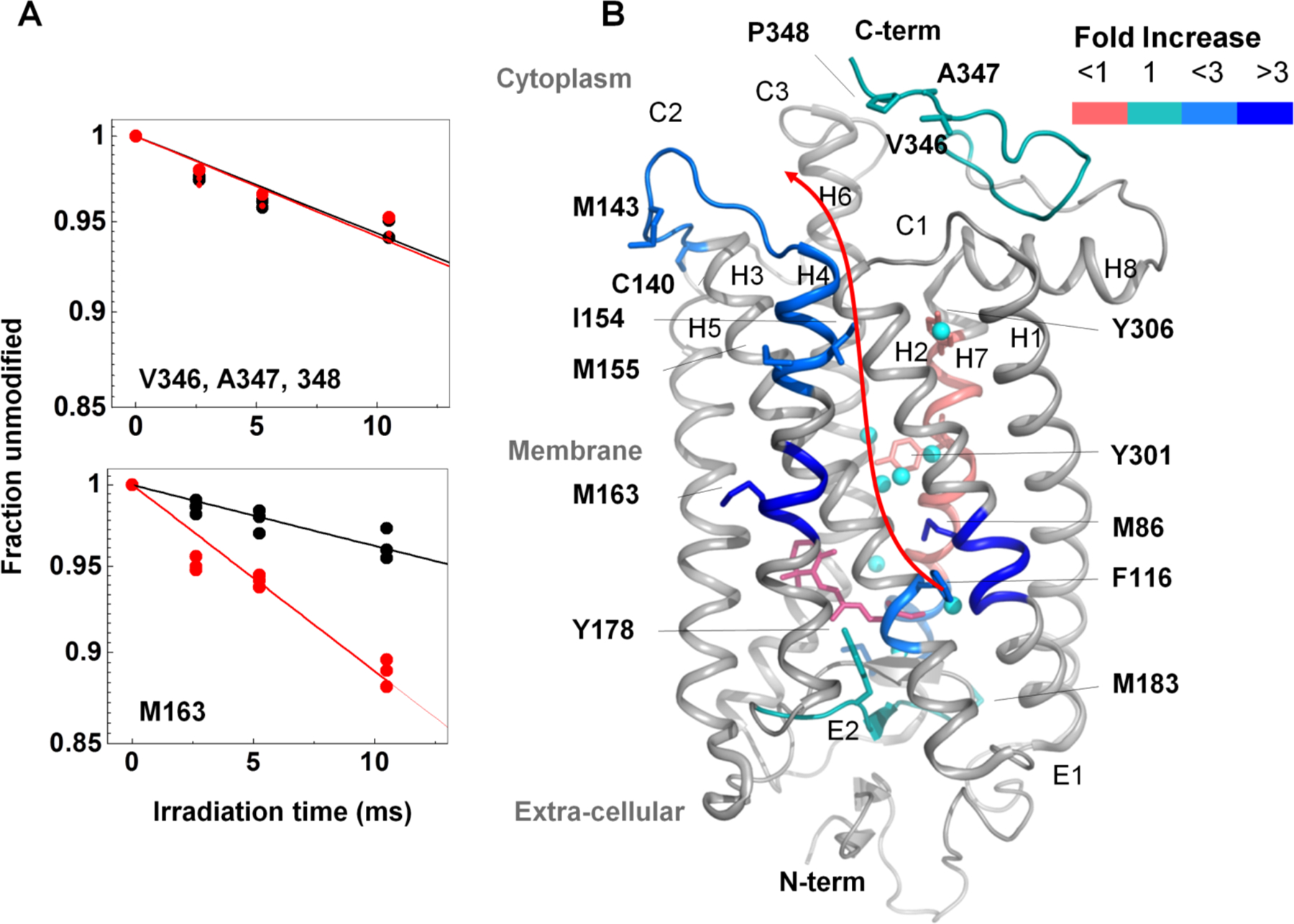
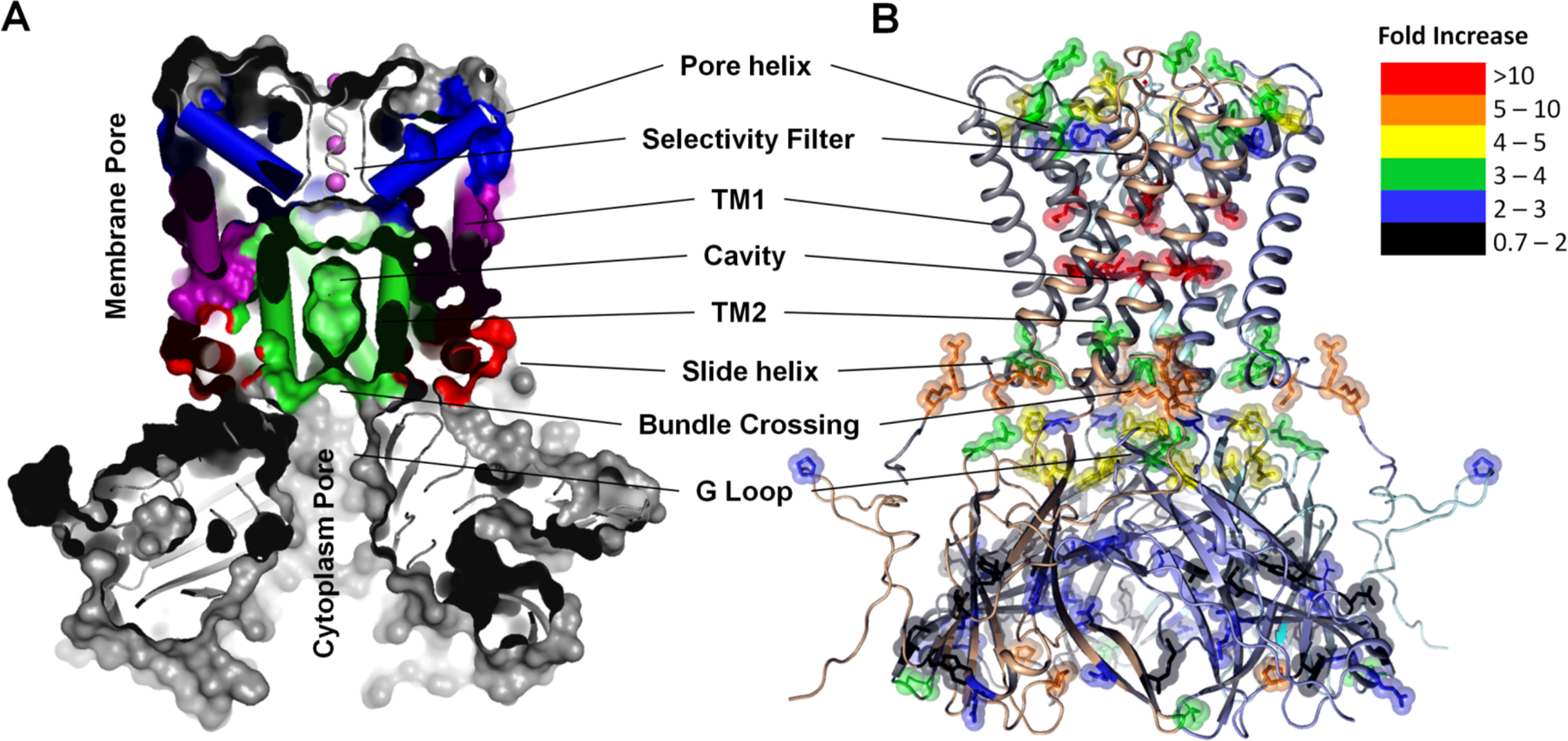
Background: Membrane proteins are crucial for cellular sensory cascades and metabolite transport, and hence are key pharmacological targets. Structural studies by traditional highresolution techniques are limited by the requirements for high purity and stability when handled in high concentration and nonnative buffers. Hence, there is a growing requirement for the use of alternate methods in a complementary but orthogonal approach to study the dynamic and functional aspects of membrane proteins in physiologically relevant conditions. In recent years, significant progress has been made in the field of X-ray radiolytic labeling in combination with mass spectroscopy, commonly known as X-ray Footprinting and Mass Spectrometry (XFMS), which provide residue-specific information on the solvent accessibility of proteins. In combination with both lowresolution biophysical methods and high-resolution structural data, XFMS is capable of providing valuable insights into structure and dynamics of membrane proteins, which have been difficult to obtain by standalone high-resolution structural techniques. The XFMS method has also demonstrated a unique capability for identification of structural waters and their dynamics in protein cavities at both a high degree of spatial and temporal resolution, and thus capable of identifying conformational hot-spots in transmembrane proteins.
Conclusion: We provide a perspective on the place of XFMS amongst other structural biology methods and showcase some of the latest developments in its usage for studying conformational changes in membrane proteins.
Keywords: Hydroxyl-radical footprinting; ion channels; mass spectrometry; oxidative labeling; radiolysis; transporters..
Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.net.
Figures
Similar articles
-
Oxidative footprinting in the study of structure and function of membrane proteins: current state and perspectives.Biochem Soc Trans. 2015 Oct;43(5):983-94. doi: 10.1042/BST20150130. Biochem Soc Trans. 2015. PMID: 26517913 Review.
-
Hydroxyl radical mediated damage of proteins in low oxygen solution investigated using X-ray footprinting mass spectrometry.J Synchrotron Radiat. 2021 Sep 1;28(Pt 5):1333-1342. doi: 10.1107/S1600577521004744. Epub 2021 Jul 20. J Synchrotron Radiat. 2021. PMID: 34475282 Free PMC article.
-
PERCEPTRON-XFMS: An Open-Source Web Server for Analysis of X-ray Footprinting with Mass Spectrometry Data.J Mol Biol. 2025 Aug 1;437(15):169127. doi: 10.1016/j.jmb.2025.169127. Epub 2025 Apr 3. J Mol Biol. 2025. PMID: 40187682
-
Recent Advances and Applications in Synchrotron X-Ray Protein Footprinting for Protein Structure and Dynamics Elucidation.Protein Pept Lett. 2016;23(3):309-22. doi: 10.2174/0929866523666160201150057. Protein Pept Lett. 2016. PMID: 26833224 Review.
-
Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization.Acc Chem Res. 2018 Mar 20;51(3):736-744. doi: 10.1021/acs.accounts.7b00593. Epub 2018 Feb 16. Acc Chem Res. 2018. PMID: 29450991 Free PMC article. Review.
Cited by
-
Connexins and Pannexins-Similarities and Differences According to the FOD-M Model.Biomedicines. 2022 Jun 25;10(7):1504. doi: 10.3390/biomedicines10071504. Biomedicines. 2022. PMID: 35884807 Free PMC article.
-
Advances in mass spectrometry-based footprinting of membrane proteins.Proteomics. 2022 Apr;22(8):e2100222. doi: 10.1002/pmic.202100222. Proteomics. 2022. PMID: 35290716 Free PMC article. Review.
-
Identification and subcellular localization analysis of membrane protein Ycf 1 in the microsporidian Nosema bombycis.PeerJ. 2022 Jul 8;10:e13530. doi: 10.7717/peerj.13530. eCollection 2022. PeerJ. 2022. PMID: 35833014 Free PMC article.
References
-
- Goldie KN; Abeyrathne P; Kebbel F; Chami M; Ringler P; Stahlberg H Cryo-electron microscopy of membrane proteins. Methods Mol. Biol, 2014, 1117, 325–341. - PubMed
