

Caspase Cleavage of Gelsolin Is an Inductive Cue for Pathologic Cardiac Hypertrophy
- PMID: 30486716
- PMCID: PMC6405540
- DOI: 10.1161/JAHA.118.010404
Caspase Cleavage of Gelsolin Is an Inductive Cue for Pathologic Cardiac Hypertrophy
Abstract
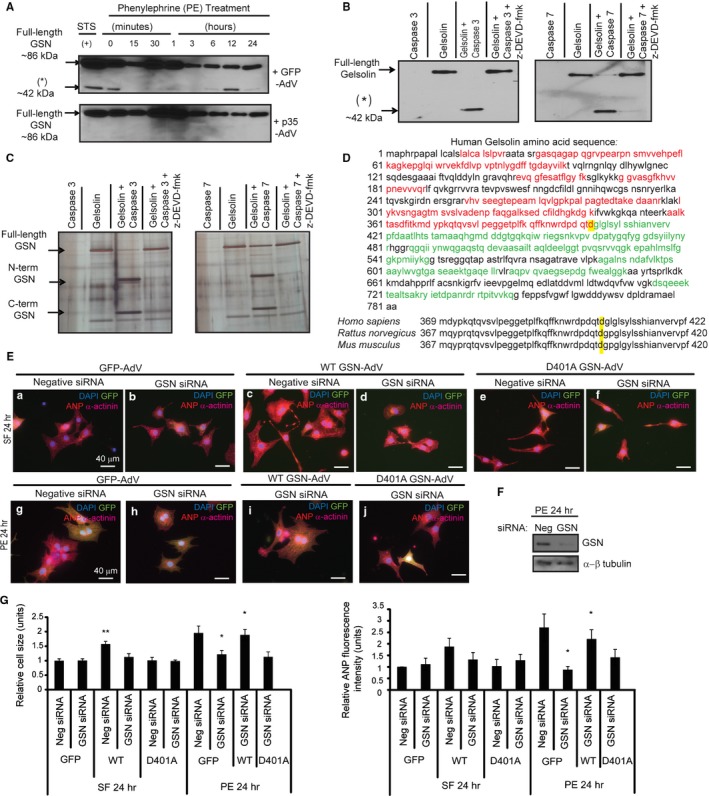
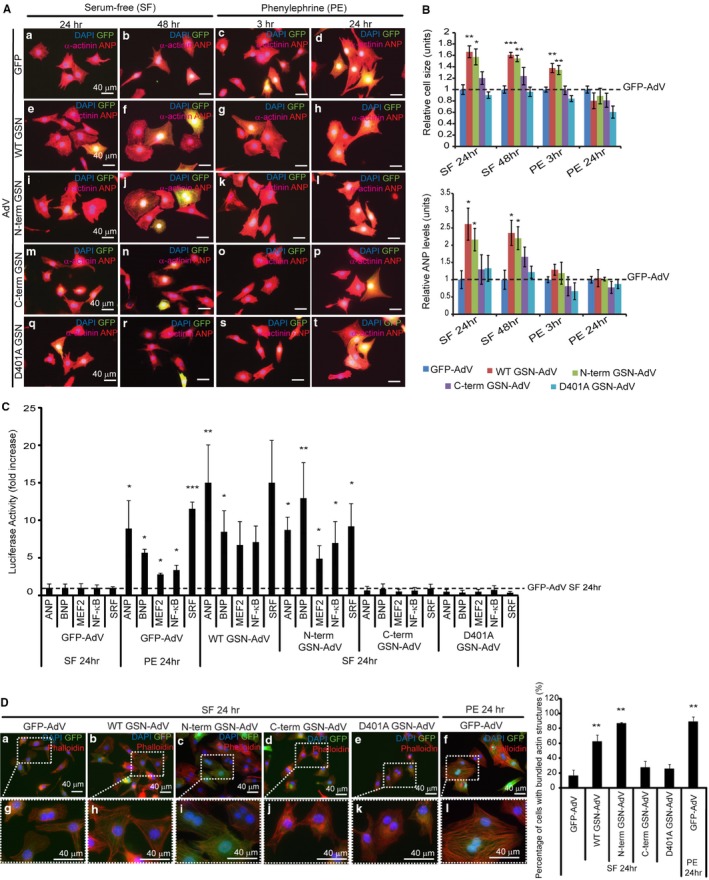
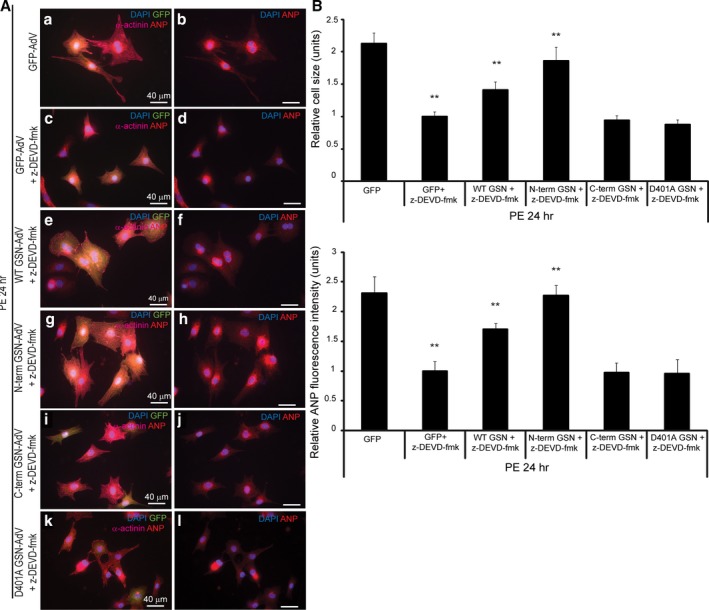
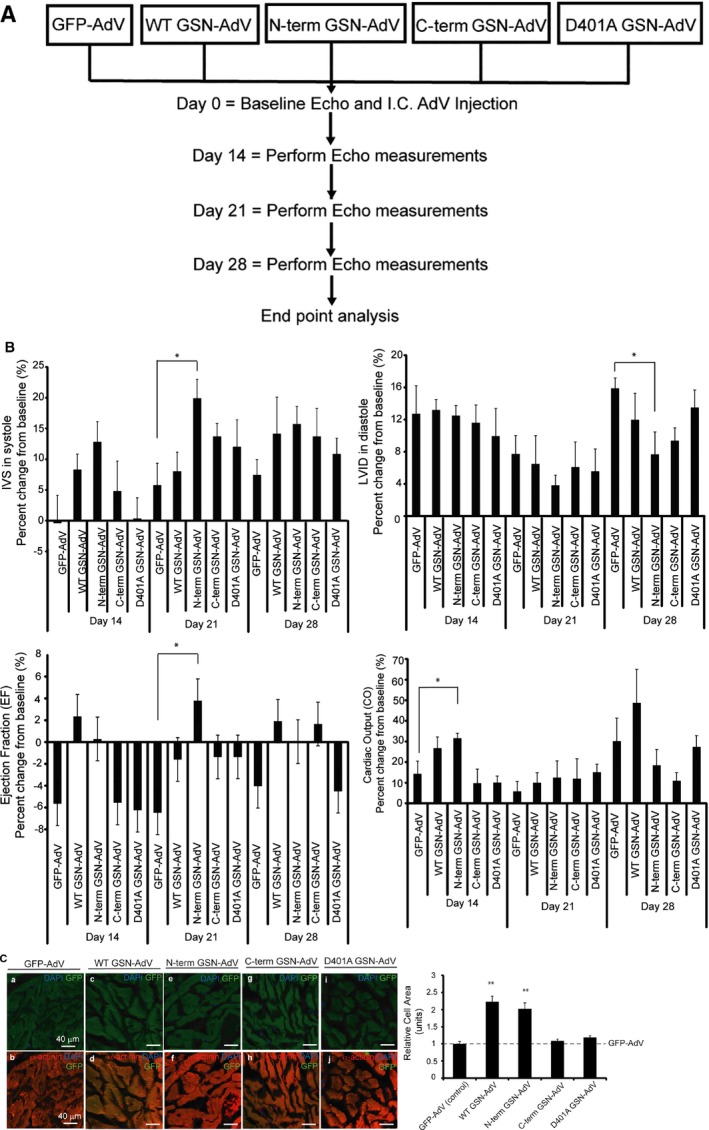
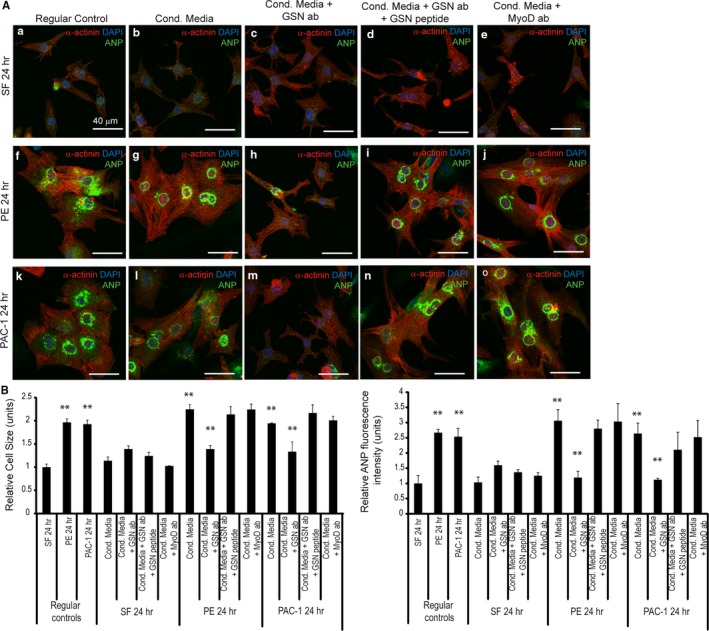
Background Cardiac hypertrophy is an adaptive remodeling event that may improve or diminish contractile performance of the heart. Physiologic and pathologic hypertrophy yield distinct outcomes, yet both are dependent on caspase-directed proteolysis. This suggests that each form of myocardial growth may derive from a specific caspase cleavage event(s). We examined whether caspase 3 cleavage of the actin capping/severing protein gelsolin is essential for the development of pathologic hypertrophy. Methods and Results Caspase targeting of gelsolin was established through protein analysis of hypertrophic cardiomyocytes and mass spectrometry mapping of cleavage sites. Pathologic agonists induced late-stage caspase-mediated cleavage of gelsolin. The requirement of caspase-mediated gelsolin cleavage for hypertrophy induction was evaluated in primary cardiomyocytes by cell size analysis, monitoring of prohypertrophy markers, and measurement of hypertrophy-related transcription activity. The in vivo impact of caspase-mediated cleavage was investigated by echo-guided intramyocardial injection of adenoviral-expressed gelsolin. Expression of the N-terminal gelsolin caspase cleavage fragment was necessary and sufficient to cause pathologic remodeling in isolated cardiomyocytes and the intact heart, whereas expression of a noncleavable form prevents cardiac remodeling. Alterations in myocardium structure and function were determined by echocardiography and end-stage cardiomyocyte cell size analysis. Gelsolin secretion was also monitored for its impact on naïve cells using competitive antibody trapping, demonstrating that hypertrophic agonist stimulation of cardiomyocytes leads to gelsolin secretion, which induces hypertrophy in naïve cells. Conclusions These results suggest that cell autonomous caspase cleavage of gelsolin is essential for pathologic hypertrophy and that cardiomyocyte secretion of gelsolin may accelerate this negative remodeling response.
Keywords: cardiac hypertrophy; cardiomyocyte hypertrophy; caspase‐3; cell signaling; cytoskeletal dynamics; gelsolin.
Figures
Similar articles
-
Intrinsic-mediated caspase activation is essential for cardiomyocyte hypertrophy.Proc Natl Acad Sci U S A. 2013 Oct 22;110(43):E4079-87. doi: 10.1073/pnas.1315587110. Epub 2013 Oct 7. Proc Natl Acad Sci U S A. 2013. PMID: 24101493 Free PMC article.
-
In vivo administration of calpeptin attenuates calpain activation and cardiomyocyte loss in pressure-overloaded feline myocardium.Am J Physiol Heart Circ Physiol. 2008 Jul;295(1):H314-26. doi: 10.1152/ajpheart.00085.2008. Epub 2008 May 16. Am J Physiol Heart Circ Physiol. 2008. PMID: 18487434 Free PMC article.
-
Gelsolin (GSN) induces cardiomyocyte hypertrophy and BNP expression via p38 signaling and GATA-4 transcriptional factor activation.Mol Cell Biochem. 2014 May;390(1-2):263-70. doi: 10.1007/s11010-014-1977-7. Epub 2014 Feb 7. Mol Cell Biochem. 2014. PMID: 24505034
-
Cardiomyocyte-specific inactivation of thyroid hormone in pathologic ventricular hypertrophy: an adaptative response or part of the problem?Heart Fail Rev. 2010 Mar;15(2):133-42. doi: 10.1007/s10741-008-9133-7. Heart Fail Rev. 2010. PMID: 19107595 Free PMC article. Review.
-
The role of Fas in the progression of ischemic heart failure: prohypertrophy or proapoptosis.Coron Artery Dis. 2008 Nov;19(7):527-34. doi: 10.1097/MCA.0b013e3283093707. Coron Artery Dis. 2008. PMID: 18923250 Review.
Cited by
-
MicroRNA205: A Key Regulator of Cardiomyocyte Transition from Proliferative to Hypertrophic Growth in the Neonatal Heart.Int J Mol Sci. 2024 Feb 12;25(4):2206. doi: 10.3390/ijms25042206. Int J Mol Sci. 2024. PMID: 38396885 Free PMC article.
-
Proteolytic activation of fatty acid synthase signals pan-stress resolution.Nat Metab. 2024 Jan;6(1):113-126. doi: 10.1038/s42255-023-00939-z. Epub 2024 Jan 2. Nat Metab. 2024. PMID: 38167727 Free PMC article.
-
Actin-Binding Proteins in Cardiac Hypertrophy.Cells. 2022 Nov 11;11(22):3566. doi: 10.3390/cells11223566. Cells. 2022. PMID: 36428995 Free PMC article. Review.
References
-
- Kong SW, Bodyak N, Yue P, Liu Z, Brown J, Izumo S, Kang PM. Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats. Physiol Genomics. 2005;21:34–42. - PubMed
-
- Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. - PubMed
-
- Dorn GW. The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
