

Developing reproducible bioinformatics analysis workflows for heterogeneous computing environments to support African genomics
- PMID: 30486782
- PMCID: PMC6264621
- DOI: 10.1186/s12859-018-2446-1
Developing reproducible bioinformatics analysis workflows for heterogeneous computing environments to support African genomics
Abstract
Background: The Pan-African bioinformatics network, H3ABioNet, comprises 27 research institutions in 17 African countries. H3ABioNet is part of the Human Health and Heredity in Africa program (H3Africa), an African-led research consortium funded by the US National Institutes of Health and the UK Wellcome Trust, aimed at using genomics to study and improve the health of Africans. A key role of H3ABioNet is to support H3Africa projects by building bioinformatics infrastructure such as portable and reproducible bioinformatics workflows for use on heterogeneous African computing environments. Processing and analysis of genomic data is an example of a big data application requiring complex interdependent data analysis workflows. Such bioinformatics workflows take the primary and secondary input data through several computationally-intensive processing steps using different software packages, where some of the outputs form inputs for other steps. Implementing scalable, reproducible, portable and easy-to-use workflows is particularly challenging.
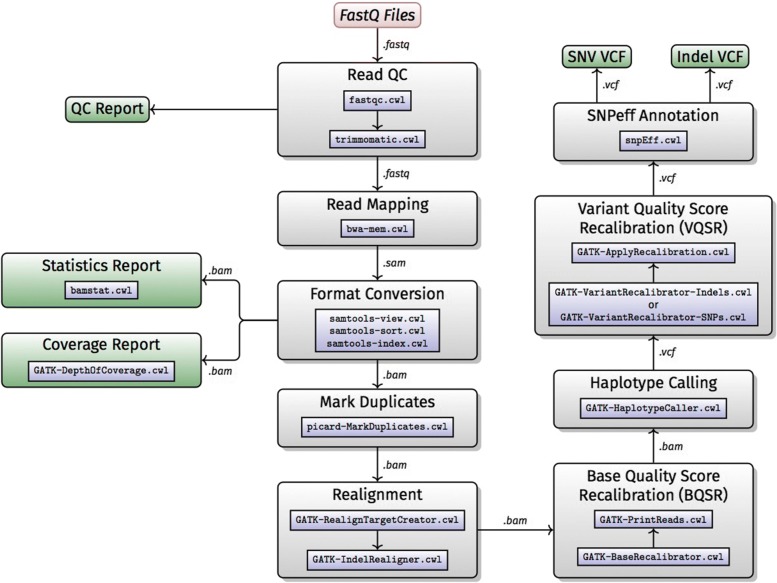
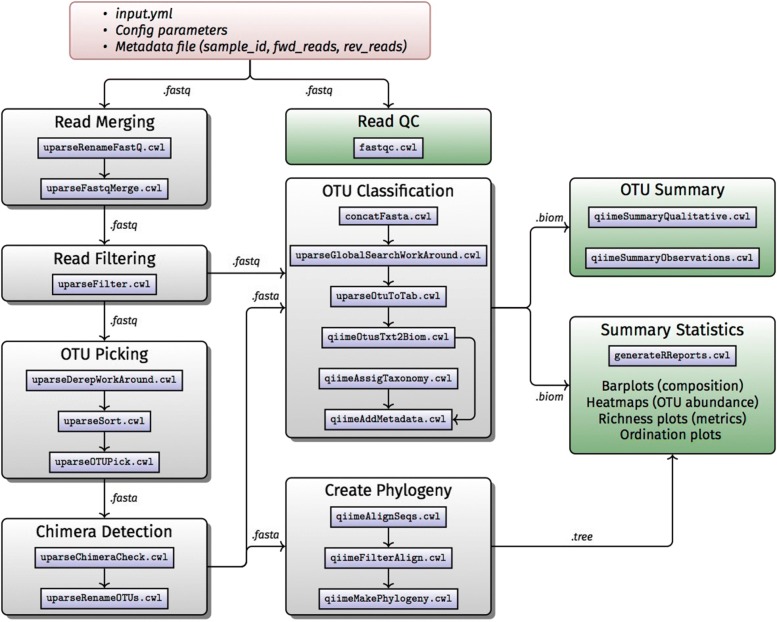
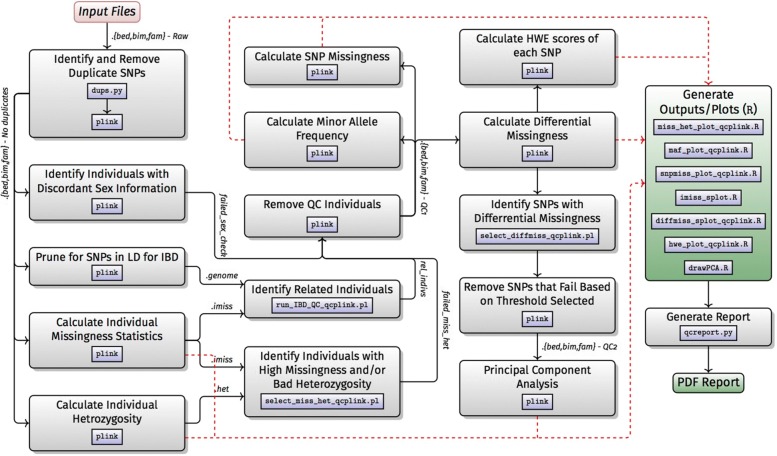
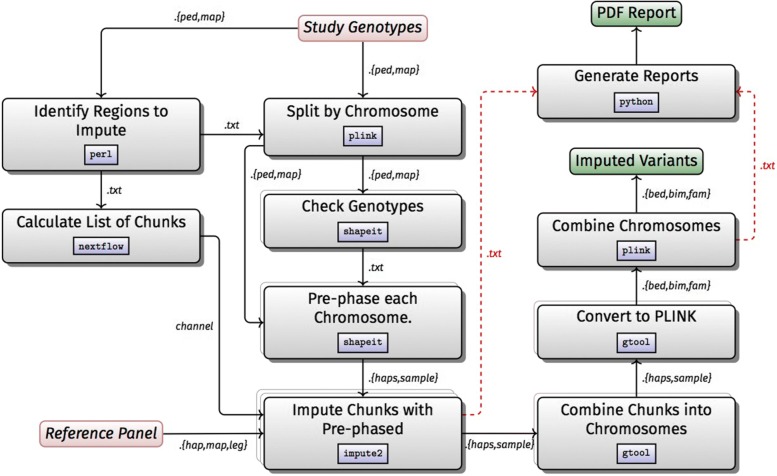
Results: H3ABioNet has built four workflows to support (1) the calling of variants from high-throughput sequencing data; (2) the analysis of microbial populations from 16S rDNA sequence data; (3) genotyping and genome-wide association studies; and (4) single nucleotide polymorphism imputation. A week-long hackathon was organized in August 2016 with participants from six African bioinformatics groups, and US and European collaborators. Two of the workflows are built using the Common Workflow Language framework (CWL) and two using Nextflow. All the workflows are containerized for improved portability and reproducibility using Docker, and are publicly available for use by members of the H3Africa consortium and the international research community.
Conclusion: The H3ABioNet workflows have been implemented in view of offering ease of use for the end user and high levels of reproducibility and portability, all while following modern state of the art bioinformatics data processing protocols. The H3ABioNet workflows will service the H3Africa consortium projects and are currently in use. All four workflows are also publicly available for research scientists worldwide to use and adapt for their respective needs. The H3ABioNet workflows will help develop bioinformatics capacity and assist genomics research within Africa and serve to increase the scientific output of H3Africa and its Pan-African Bioinformatics Network.
Keywords: Africa; Bioinformatics; Docker; Genomics; Pipeline; Reproducibility; Workflows.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Michael R. Crusoe, in his role as CWL Community Engineer, has had his salary supported in the past by grants from Seven Bridges Genomics, Inc to his employers. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Liu Bo, Madduri Ravi K, Sotomayor Borja, Chard Kyle, Lacinski Lukasz, Dave Utpal J, Li Jianqiang, Liu Chunchen, Foster Ian T. Cloud-based bioinformatics workflow platform for large-scale next-generation sequencing analyses. Journal of Biomedical Informatics. 2014;49:119–133. doi: 10.1016/j.jbi.2014.01.005. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
