

Clinical potential of mass spectrometry-based proteogenomics
- PMID: 30487530
- PMCID: PMC6448780
- DOI: 10.1038/s41571-018-0135-7
Clinical potential of mass spectrometry-based proteogenomics
Abstract
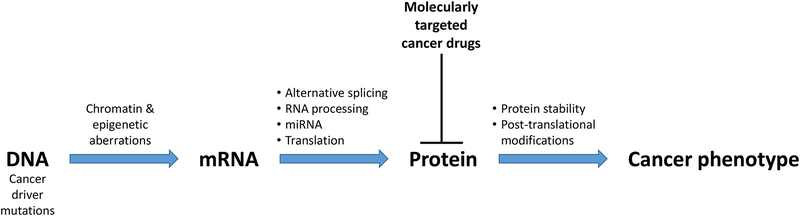
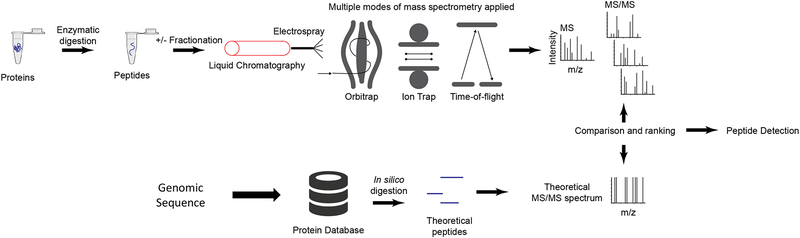
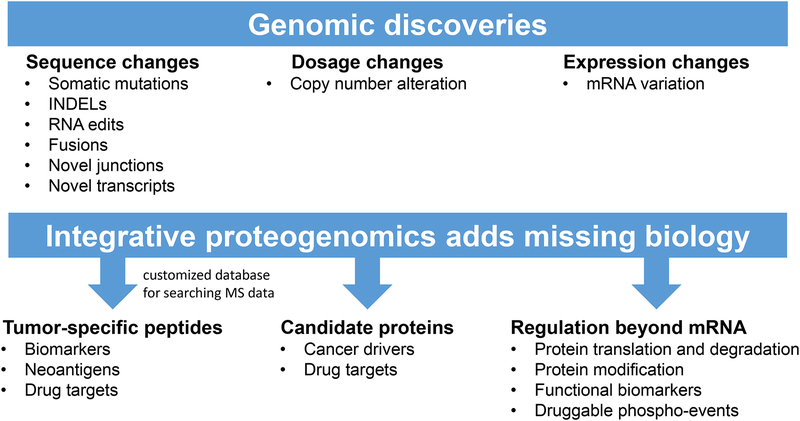
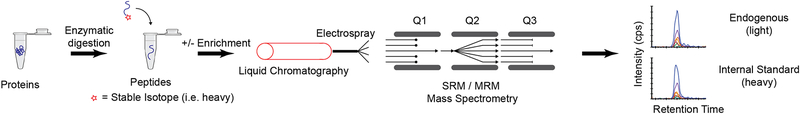
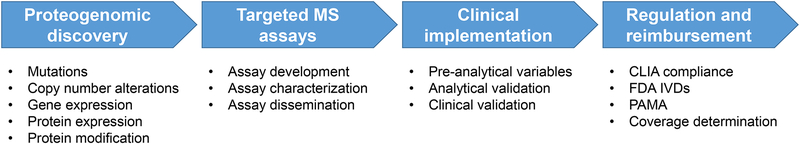
Cancer genomics research aims to advance personalized oncology by finding and targeting specific genetic alterations associated with cancers. In genome-driven oncology, treatments are selected for individual patients on the basis of the findings of tumour genome sequencing. This personalized approach has prolonged the survival of subsets of patients with cancer. However, many patients do not respond to the predicted therapies based on the genomic profiles of their tumours. Furthermore, studies pairing genomic and proteomic analyses of samples from the same tumours have shown that the proteome contains novel information that cannot be discerned through genomic analysis alone. This observation has led to the concept of proteogenomics, in which both types of data are leveraged for a more complete view of tumour biology that might enable patients to be more successfully matched to effective treatments than they would using genomics alone. In this Perspective, we discuss the added value of proteogenomics over the current genome-driven approach to the clinical characterization of cancers and summarize current efforts to incorporate targeted proteomic measurements based on selected/multiple reaction monitoring (SRM/MRM) mass spectrometry into the clinical laboratory to facilitate clinical proteogenomics.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
