

Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis
- PMID: 30487573
- PMCID: PMC6261936
- DOI: 10.1038/s41467-018-07368-7
Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis
Abstract
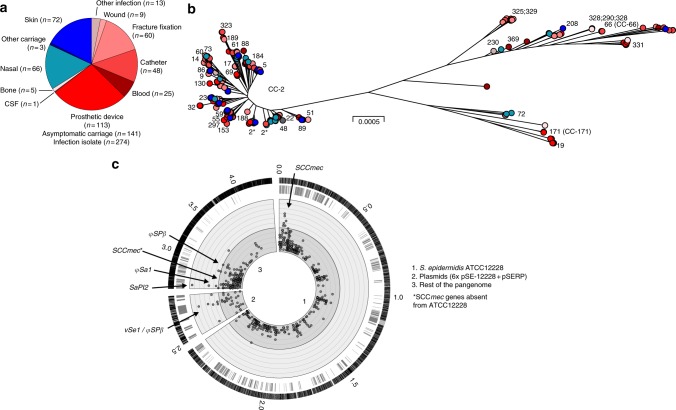
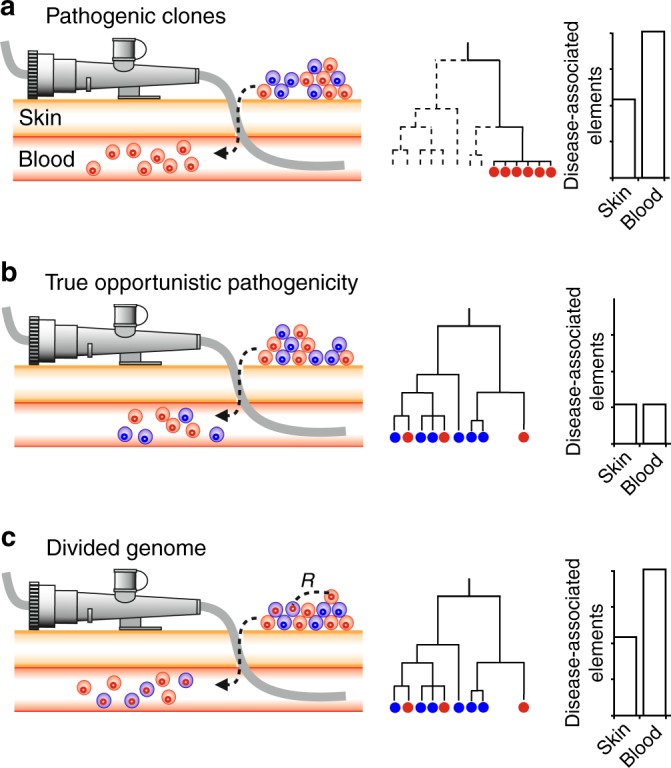
Some of the most common infectious diseases are caused by bacteria that naturally colonise humans asymptomatically. Combating these opportunistic pathogens requires an understanding of the traits that differentiate infecting strains from harmless relatives. Staphylococcus epidermidis is carried asymptomatically on the skin and mucous membranes of virtually all humans but is a major cause of nosocomial infection associated with invasive procedures. Here we address the underlying evolutionary mechanisms of opportunistic pathogenicity by combining pangenome-wide association studies and laboratory microbiology to compare S. epidermidis from bloodstream and wound infections and asymptomatic carriage. We identify 61 genes containing infection-associated genetic elements (k-mers) that correlate with in vitro variation in known pathogenicity traits (biofilm formation, cell toxicity, interleukin-8 production, methicillin resistance). Horizontal gene transfer spreads these elements, allowing divergent clones to cause infection. Finally, Random Forest model prediction of disease status (carriage vs. infection) identifies pathogenicity elements in 415 S. epidermidis isolates with 80% accuracy, demonstrating the potential for identifying risk genotypes pre-operatively.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Weinstein MP, et al. The clinical significance of positive blood cultures in the 1990s: a prospective comprehensive evaluation of the microbiology, epidemiology, and outcome of bacteremia and fungemia in adults. Clin. Infect. Dis.: Off. Publ. Infect. Dis. Soc. Am. 1997;24:584–602. doi: 10.1093/clind/24.4.584. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
