

CDK12 regulates DNA repair genes by suppressing intronic polyadenylation
- PMID: 30487607
- PMCID: PMC6328294
- DOI: 10.1038/s41586-018-0758-y
CDK12 regulates DNA repair genes by suppressing intronic polyadenylation
Abstract
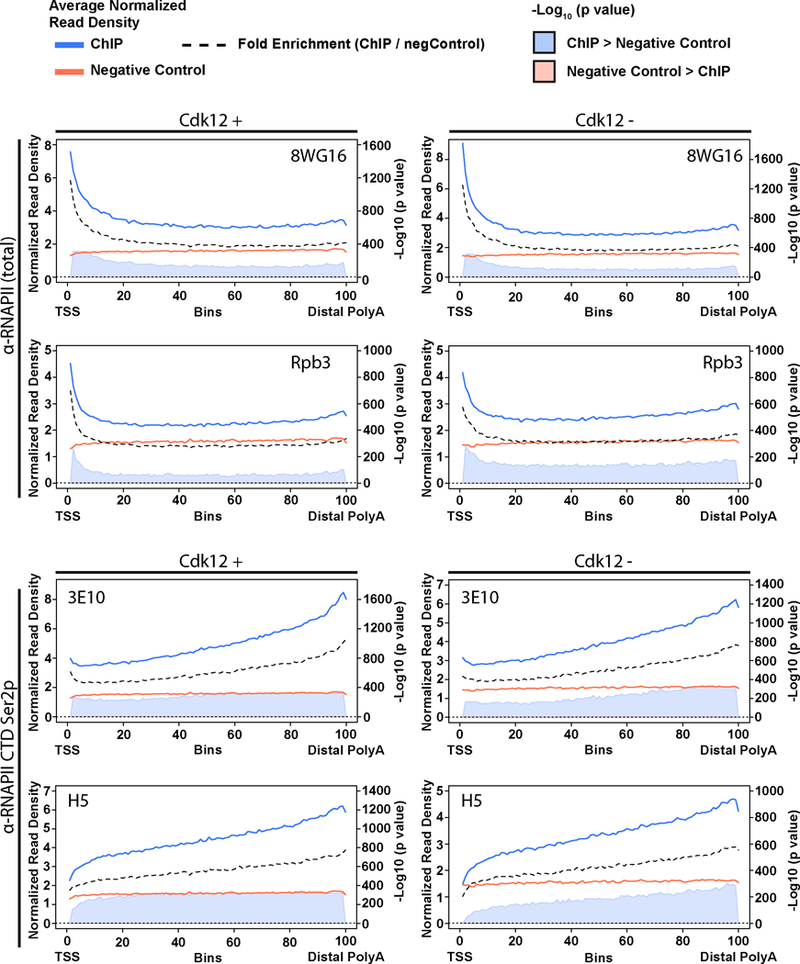
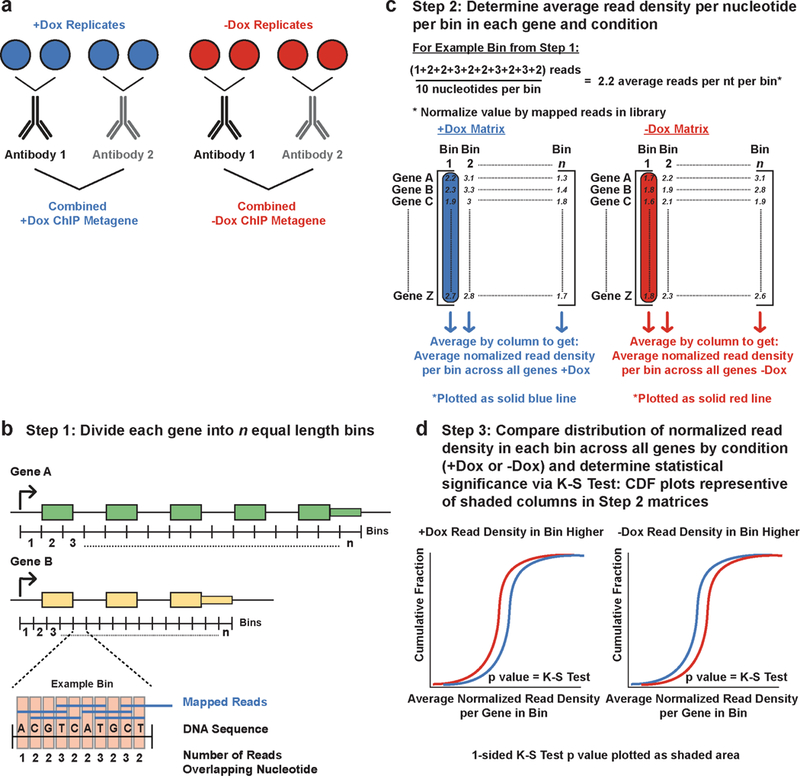
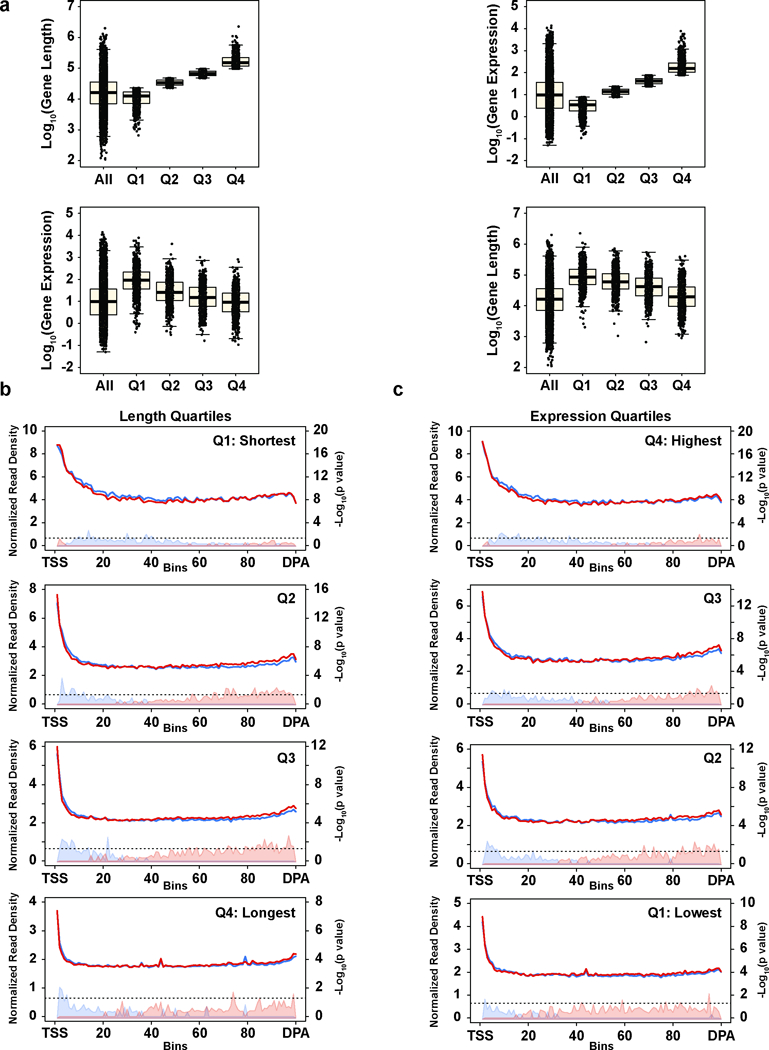
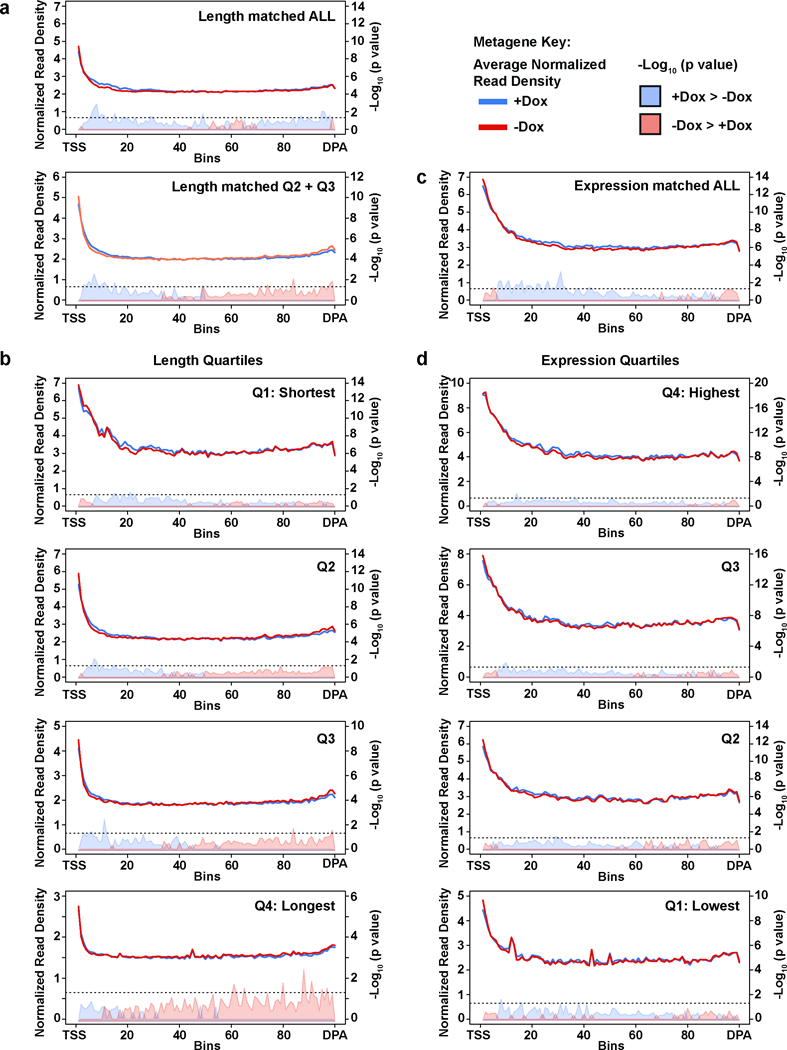
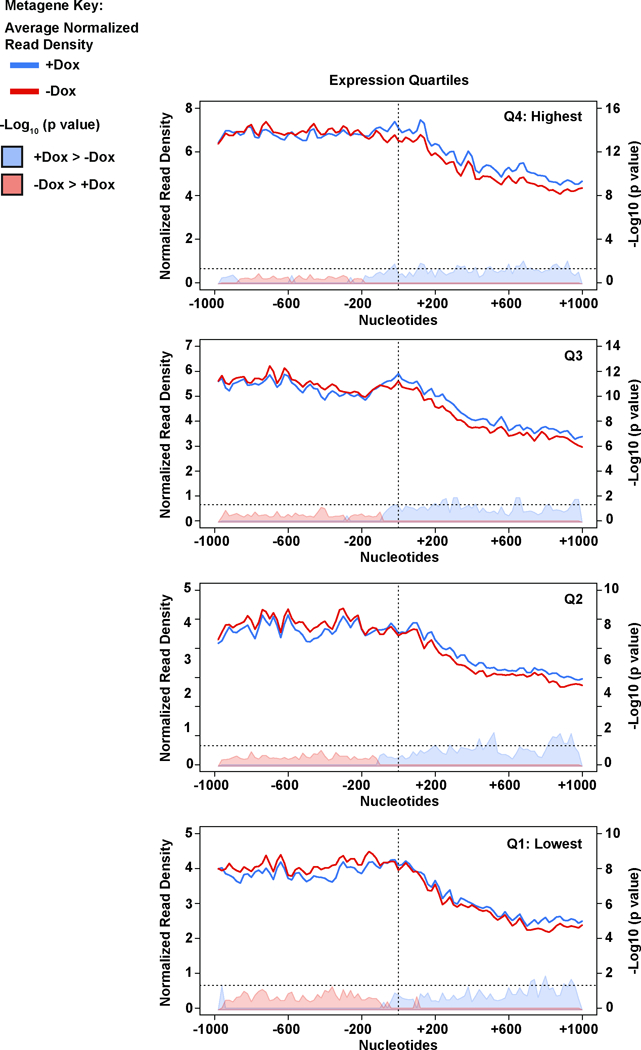
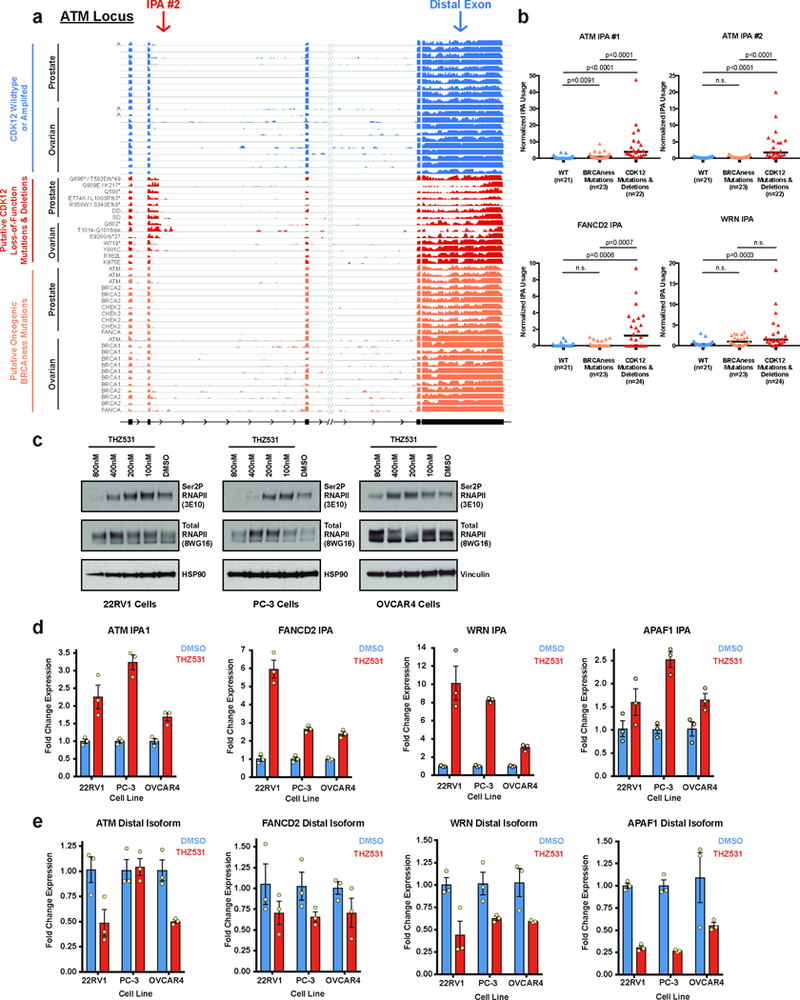
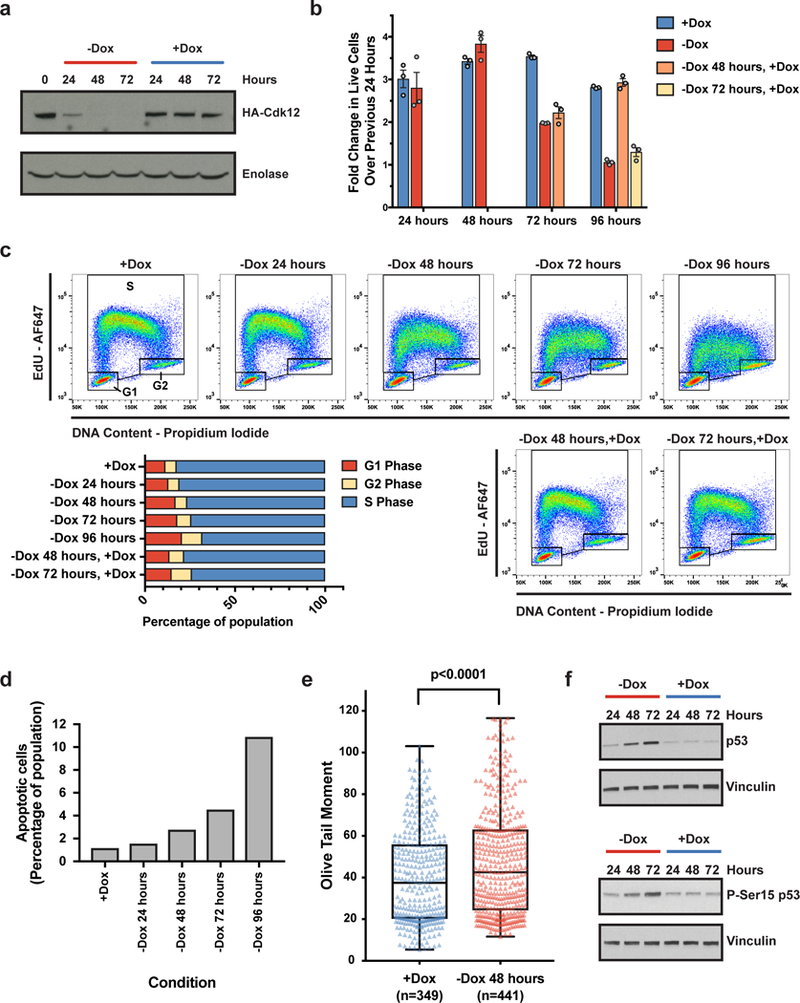
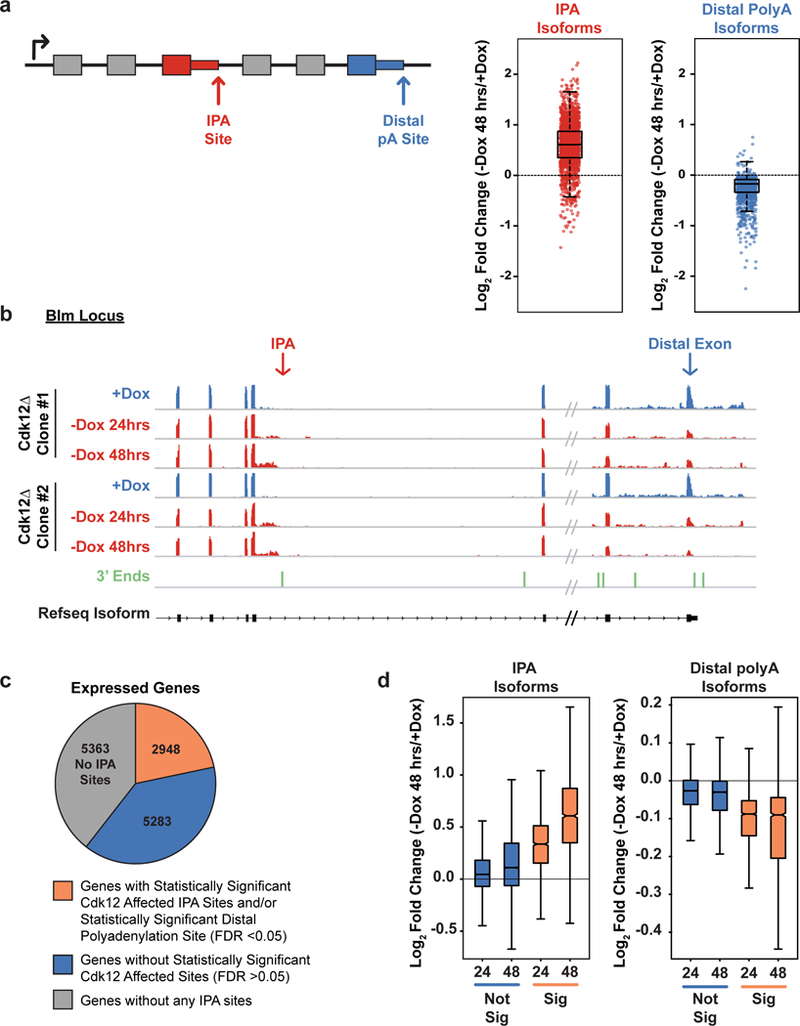
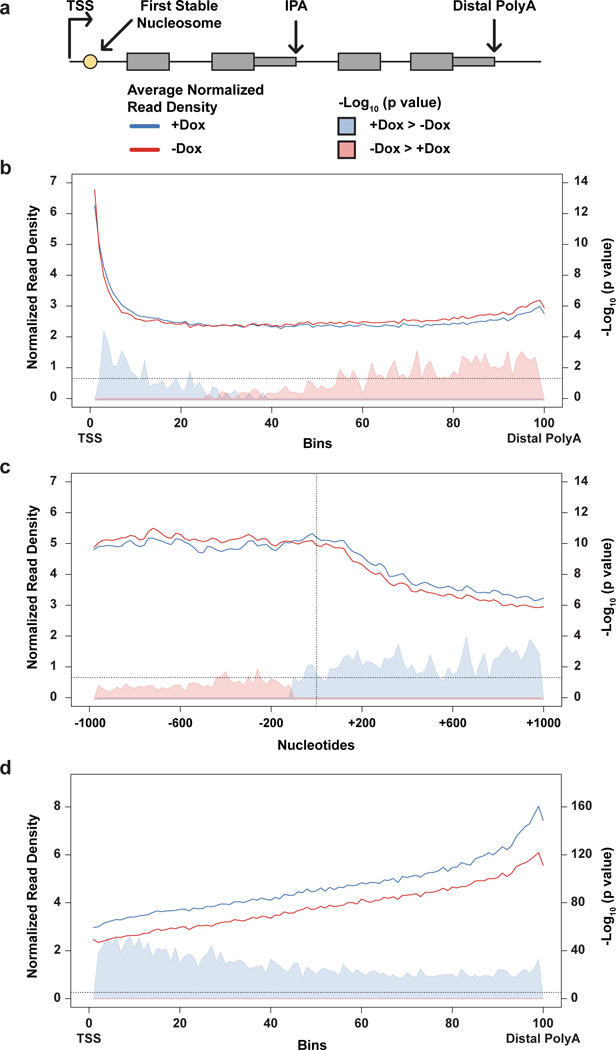
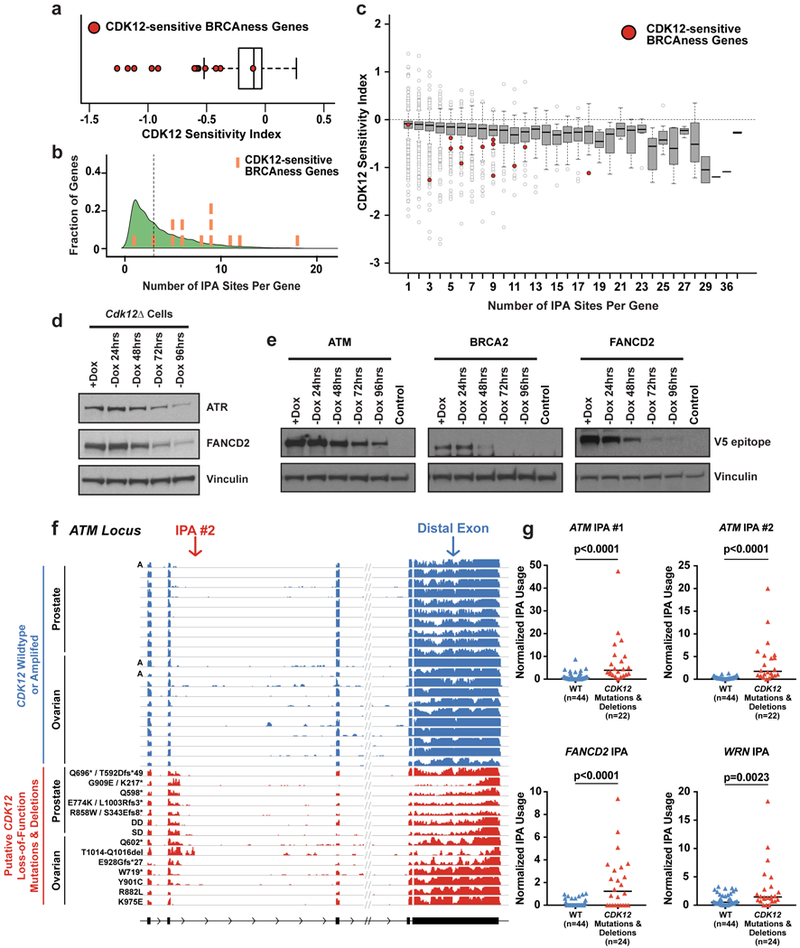
Mutations that attenuate homologous recombination (HR)-mediated repair promote tumorigenesis and sensitize cells to chemotherapeutics that cause replication fork collapse, a phenotype known as 'BRCAness'1. BRCAness tumours arise from loss-of-function mutations in 22 genes1. Of these genes, all but one (CDK12) function directly in the HR repair pathway1. CDK12 phosphorylates serine 2 of the RNA polymerase II C-terminal domain heptapeptide repeat2-7, a modification that regulates transcription elongation, splicing, and cleavage and polyadenylation8,9. Genome-wide expression studies suggest that depletion of CDK12 abrogates the expression of several HR genes relatively specifically, thereby blunting HR repair3-7,10,11. This observation suggests that the mutational status of CDK12 may predict sensitivity to targeted treatments against BRCAness, such as PARP1 inhibitors, and that CDK12 inhibitors may induce sensitization of HR-competent tumours to these treatments6,7,10,11. Despite growing clinical interest, the mechanism by which CDK12 regulates HR genes remains unknown. Here we show that CDK12 globally suppresses intronic polyadenylation events in mouse embryonic stem cells, enabling the production of full-length gene products. Many HR genes harbour more intronic polyadenylation sites than other expressed genes, and these sites are particularly sensitive to loss of CDK12. The cumulative effect of these sites accounts for the enhanced sensitivity of HR gene expression to CDK12 loss, and we find that this mechanism is conserved in human tumours that contain loss-of-function CDK12 mutations. This work clarifies the function of CDK12 and underscores its potential both as a chemotherapeutic target and as a tumour biomarker.
Figures




Comment in
-
A Link between Intronic Polyadenylation and HR Maintenance Discovered.Biochemistry. 2019 Apr 9;58(14):1835-1836. doi: 10.1021/acs.biochem.9b00202. Epub 2019 Mar 28. Biochemistry. 2019. PMID: 30920804 No abstract available.
References
-
- Lord CJ & Ashworth A BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016). - PubMed
Additional References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
