

The Pseudomonas aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity
- PMID: 30496396
- PMCID: PMC6328365
- DOI: 10.1093/gbe/evy259
The Pseudomonas aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity
Abstract
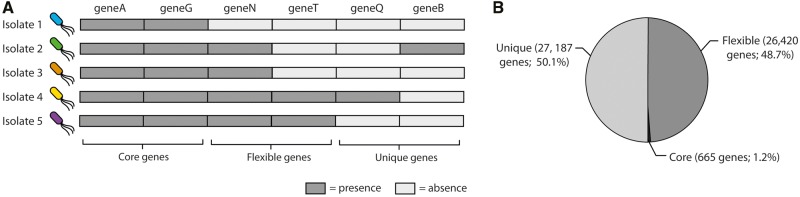
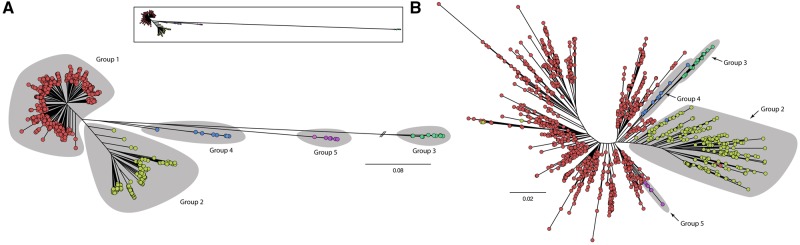
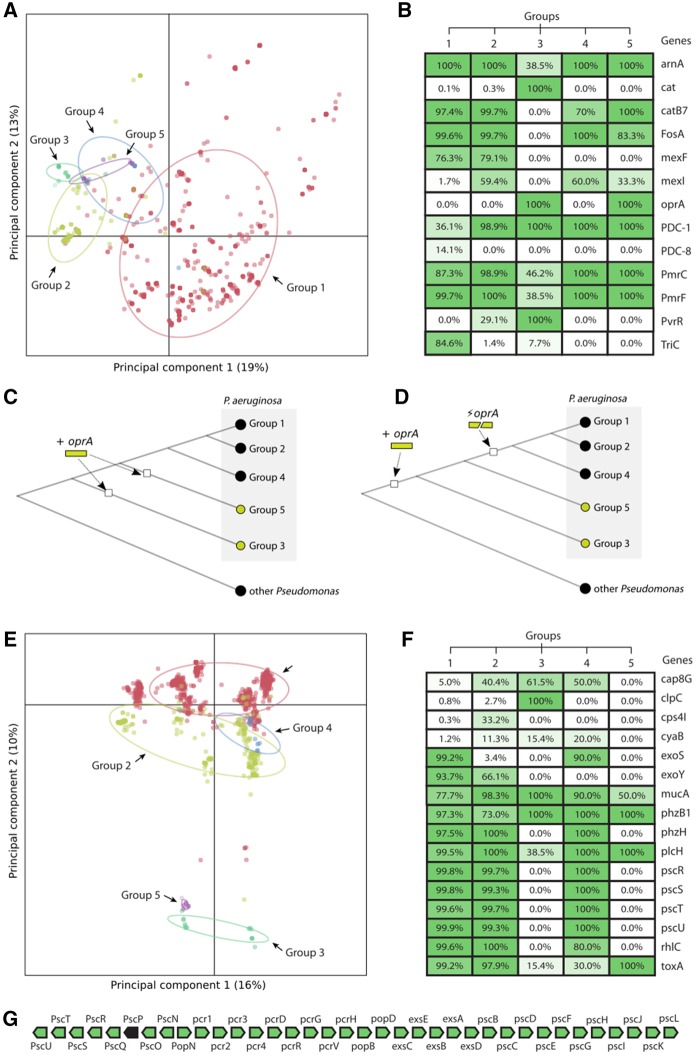
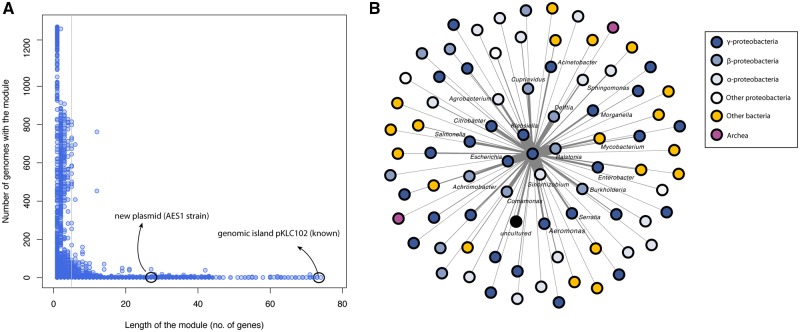
The huge increase in the availability of bacterial genomes led us to a point in which we can investigate and query pan-genomes, for example, the full set of genes of a given bacterial species or clade. Here, we used a data set of 1,311 high-quality genomes from the human pathogen Pseudomonas aeruginosa, 619 of which were newly sequenced, to show that a pan-genomic approach can greatly refine the population structure of bacterial species, provide new insights to define species boundaries, and generate hypotheses on the evolution of pathogenicity. The 665-gene P. aeruginosa core genome presented here, which constitutes only 1% of the entire pan-genome, is the first to be in the same order of magnitude as the minimal bacterial genome and represents a conservative estimate of the actual core genome. Moreover, the phylogeny based on this core genome provides strong evidence for a five-group population structure that includes two previously undescribed groups of isolates. Comparative genomics focusing on antimicrobial resistance and virulence genes showed that variation among isolates was partly linked to this population structure. Finally, we hypothesized that horizontal gene transfer had an important role in this respect, and found a total of 3,010 putative complete and fragmented plasmids, 5% and 12% of which contained resistance or virulence genes, respectively. This work provides data and strategies to study the evolutionary trajectories of resistance and virulence in P. aeruginosa.
Figures
References
-
- Bodey GP, Bolivar R, Fainstein V, Jadeja L. 1983. Infections caused by Pseudomonas aeruginosa. Rev Infect Dis. 5(2):279–313. - PubMed
-
- Dray S, Dufour A. 2007. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 22:1–20.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
