

Recovery and analysis of transcriptome subsets from pooled single-cell RNA-seq libraries
- PMID: 30496484
- PMCID: PMC6393243
- DOI: 10.1093/nar/gky1204
Recovery and analysis of transcriptome subsets from pooled single-cell RNA-seq libraries
Abstract
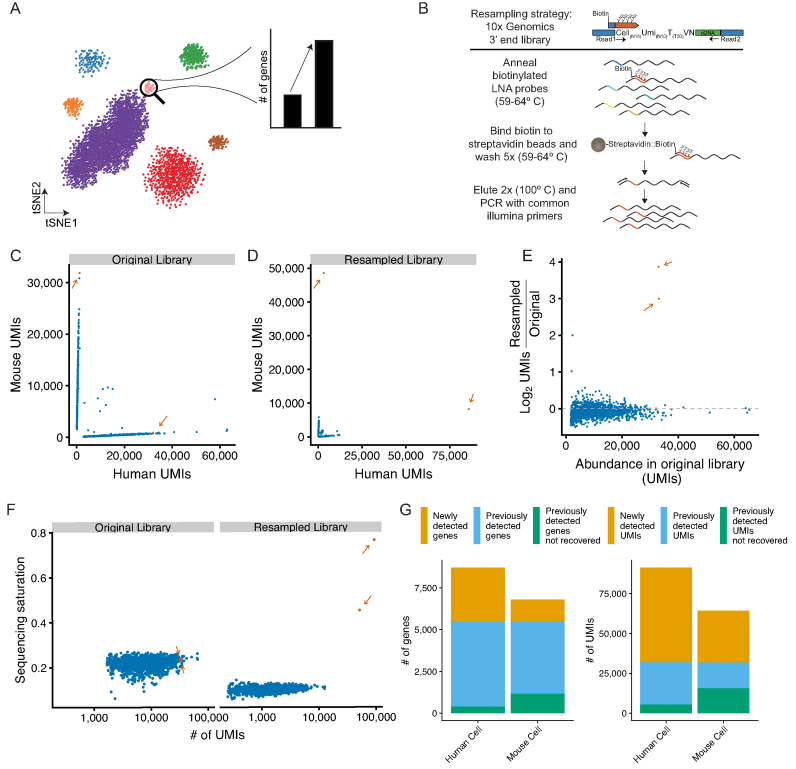
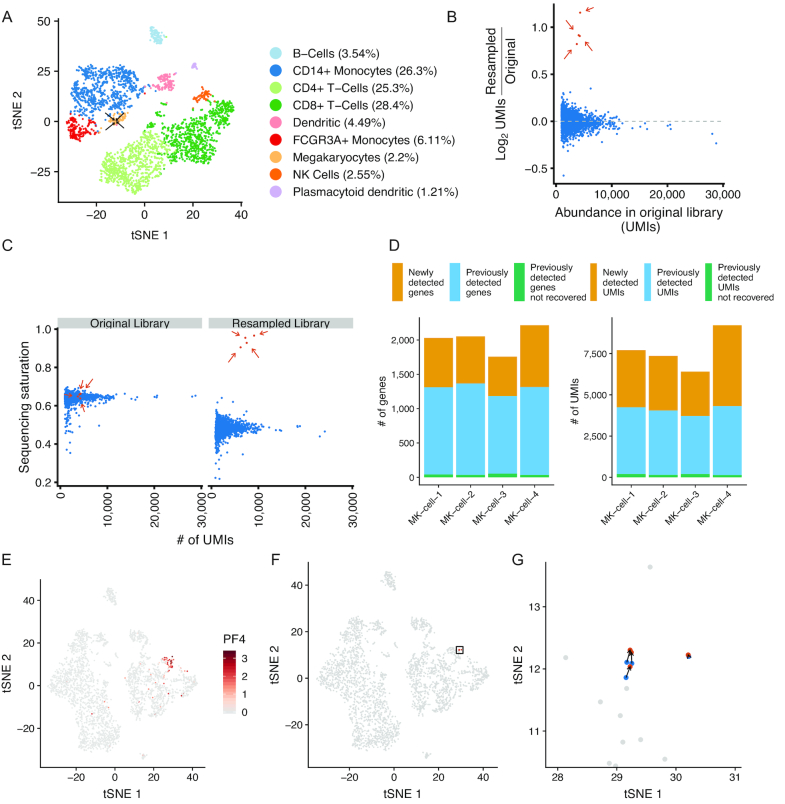
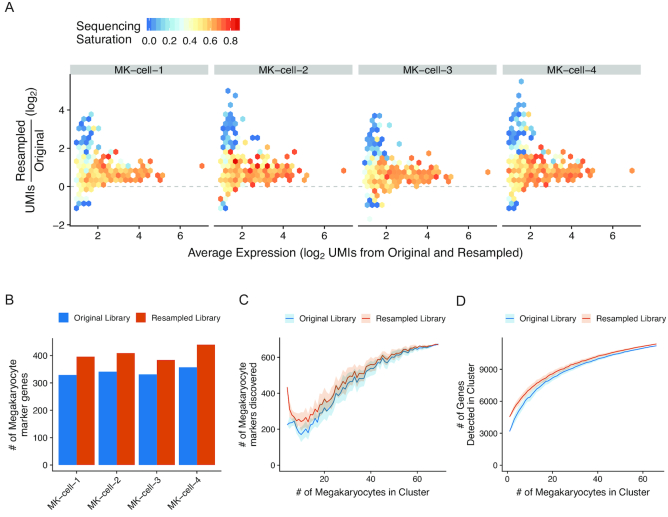
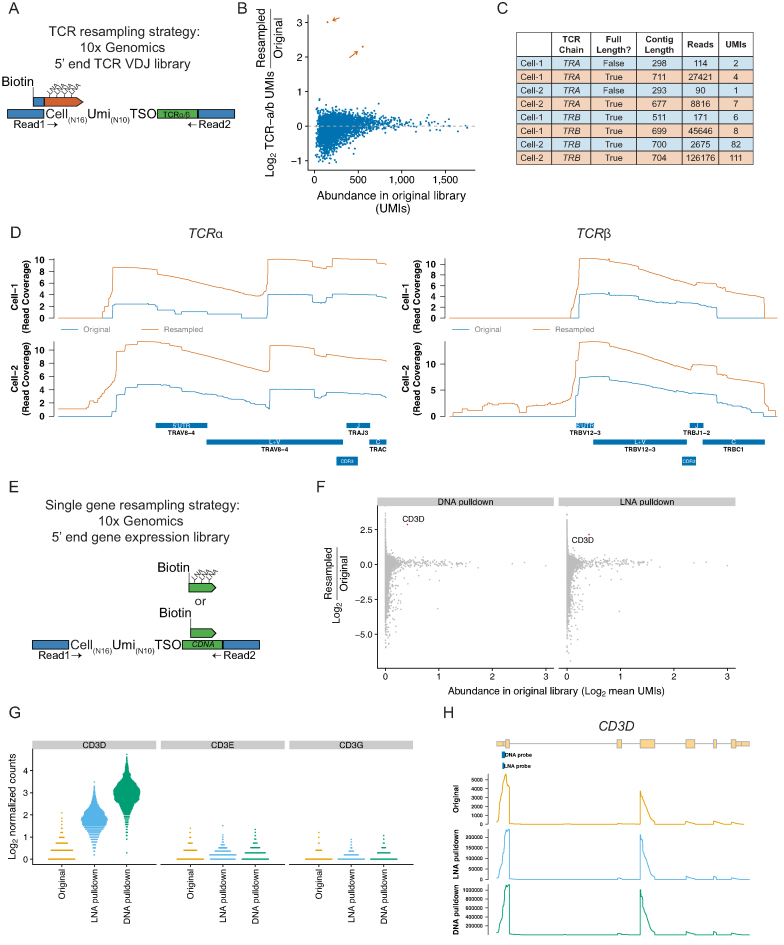
Single-cell RNA sequencing (scRNA-seq) methods generate sparse gene expression profiles for thousands of single cells in a single experiment. The information in these profiles is sufficient to classify cell types by distinct expression patterns but the high complexity of scRNA-seq libraries often prevents full characterization of transcriptomes from individual cells. To extract more focused gene expression information from scRNA-seq libraries, we developed a strategy to physically recover the DNA molecules comprising transcriptome subsets, enabling deeper interrogation of the isolated molecules by another round of DNA sequencing. We applied the method in cell-centric and gene-centric modes to isolate cDNA fragments from scRNA-seq libraries. First, we resampled the transcriptomes of rare, single megakaryocytes from a complex mixture of lymphocytes and analyzed them in a second round of DNA sequencing, yielding up to 20-fold greater sequencing depth per cell and increasing the number of genes detected per cell from a median of 1313 to 2002. We similarly isolated mRNAs from targeted T cells to improve the reconstruction of their VDJ-rearranged immune receptor mRNAs. Second, we isolated CD3D mRNA fragments expressed across cells in a scRNA-seq library prepared from a clonal T cell line, increasing the number of cells with detected CD3D expression from 59.7% to 100%. Transcriptome resampling is a general approach to recover targeted gene expression information from single-cell RNA sequencing libraries that enhances the utility of these costly experiments, and may be applicable to the targeted recovery of molecules from other single-cell assays.
© The Author(s) 2018. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Pollen A.A., Nowakowski T.J., Shuga J., Wang X., Leyrat A.A., Lui J.H., Li N., Szpankowski L., Fowler B., Chen P. et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014; 32:1053–1058. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
