

In vivo induction of membrane damage by β-amyloid peptide oligomers
- PMID: 30497524
- PMCID: PMC6263551
- DOI: 10.1186/s40478-018-0634-x
In vivo induction of membrane damage by β-amyloid peptide oligomers
Abstract
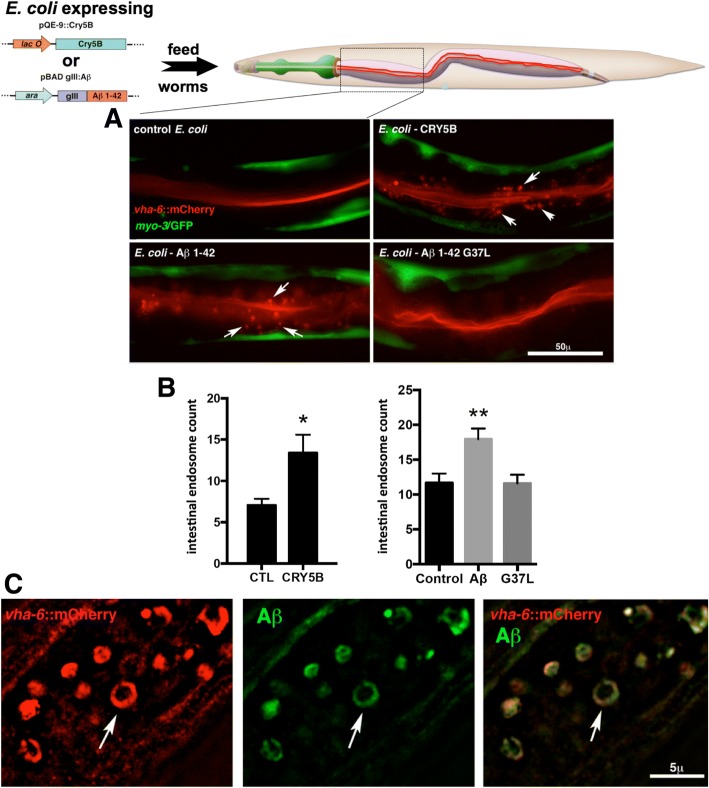
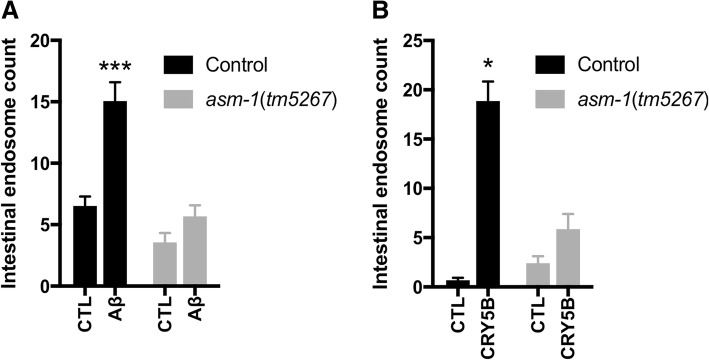
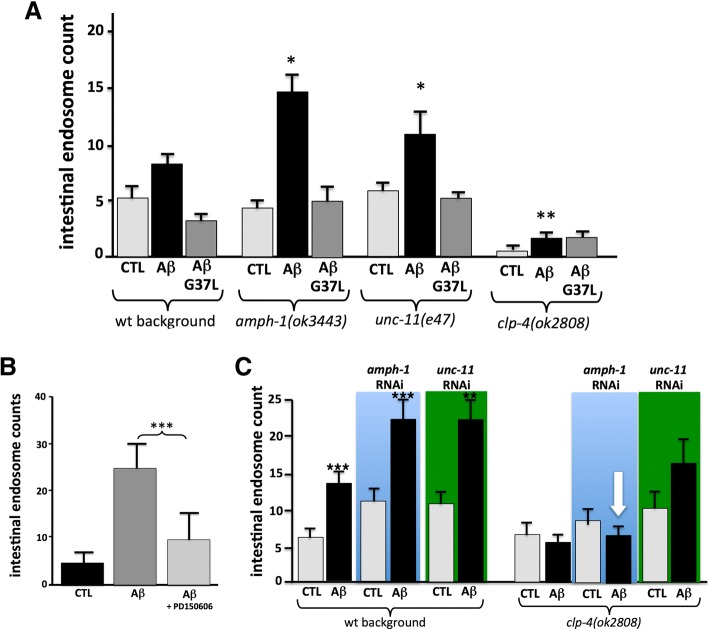
Exposure to the β-amyloid peptide (Aβ) is toxic to neurons and other cell types, but the mechanism(s) involved are still unresolved. Synthetic Aβ oligomers can induce ion-permeable pores in synthetic membranes, but whether this ability to damage membranes plays a role in the ability of Aβ oligomers to induce tau hyperphosphorylation, or other disease-relevant pathological changes, is unclear. To examine the cellular responses to Aβ exposure independent of possible receptor interactions, we have developed an in vivo C. elegans model that allows us to visualize these cellular responses in living animals. We find that feeding C. elegans E. coli expressing human Aβ induces a membrane repair response similar to that induced by exposure to the CRY5B, a known pore-forming toxin produced by B. thuringensis. This repair response does not occur when C. elegans is exposed to an Aβ Gly37Leu variant, which we have previously shown to be incapable of inducing tau phosphorylation in hippocampal neurons. The repair response is also blocked by loss of calpain function, and is altered by loss-of-function mutations in the C. elegans orthologs of BIN1 and PICALM, well-established risk genes for late onset Alzheimer's disease. To investigate the role of membrane repair on tau phosphorylation directly, we exposed hippocampal neurons to streptolysin O (SLO), a pore-forming toxin that induces a well-characterized membrane repair response. We find that SLO induces tau hyperphosphorylation, which is blocked by calpain inhibition. Finally, we use a novel biarsenical dye-tagging approach to show that the Gly37Leu substitution interferes with Aβ multimerization and thus the formation of potentially pore-forming oligomers. We propose that Aβ-induced tau hyperphosphorylation may be a downstream consequence of induction of a membrane repair process.
Keywords: Alzheimer’s disease; Caenorhabditis elegans; Pore-forming toxin; Tau; β-amyloid.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures





Similar articles
-
The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons.Neurochem Int. 2008 Sep;53(3-4):79-88. doi: 10.1016/j.neuint.2008.06.003. Epub 2008 Jun 12. Neurochem Int. 2008. PMID: 18590784 Free PMC article.
-
eEF2K inhibition blocks Aβ42 neurotoxicity by promoting an NRF2 antioxidant response.Acta Neuropathol. 2017 Jan;133(1):101-119. doi: 10.1007/s00401-016-1634-1. Epub 2016 Oct 17. Acta Neuropathol. 2017. PMID: 27752775
-
Humanin Specifically Interacts with Amyloid-β Oligomers and Counteracts Their in vivo Toxicity.J Alzheimers Dis. 2017;57(3):857-871. doi: 10.3233/JAD-160951. J Alzheimers Dis. 2017. PMID: 28282805
-
Alzheimer's disease.Subcell Biochem. 2012;65:329-52. doi: 10.1007/978-94-007-5416-4_14. Subcell Biochem. 2012. PMID: 23225010 Review.
-
Understanding the molecular basis of Alzheimer's disease using a Caenorhabditis elegans model system.Brain Struct Funct. 2010 Mar;214(2-3):263-83. doi: 10.1007/s00429-009-0235-3. Epub 2009 Dec 11. Brain Struct Funct. 2010. PMID: 20012092 Free PMC article. Review.
Cited by
-
Exogenous misfolded protein oligomers can cross the intestinal barrier and cause a disease phenotype in C. elegans.Sci Rep. 2021 Jul 13;11(1):14391. doi: 10.1038/s41598-021-93527-8. Sci Rep. 2021. PMID: 34257326 Free PMC article.
-
Amyloid-β oligomers increase the binding and internalization of tau oligomers in human synapses.Acta Neuropathol. 2024 Dec 17;149(1):2. doi: 10.1007/s00401-024-02839-2. Acta Neuropathol. 2024. PMID: 39688618 Free PMC article.
-
Prediction of Transmembrane Regions, Cholesterol, and Ganglioside Binding Sites in Amyloid-Forming Proteins Indicate Potential for Amyloid Pore Formation.Front Mol Neurosci. 2021 Feb 10;14:619496. doi: 10.3389/fnmol.2021.619496. eCollection 2021. Front Mol Neurosci. 2021. PMID: 33642992 Free PMC article.
-
Cholesterol as a key player in amyloid β-mediated toxicity in Alzheimer's disease.Front Mol Neurosci. 2022 Aug 25;15:937056. doi: 10.3389/fnmol.2022.937056. eCollection 2022. Front Mol Neurosci. 2022. PMID: 36090253 Free PMC article. Review.
-
Discovery of Selective Butyrylcholinesterase (BChE) Inhibitors through a Combination of Computational Studies and Biological Evaluations.Molecules. 2019 Nov 20;24(23):4217. doi: 10.3390/molecules24234217. Molecules. 2019. PMID: 31757047 Free PMC article.
References
-
- Alberdi E, Sanchez-Gomez MV, Cavaliere F, Perez-Samartin A, Zugaza JL, Trullas R, Domercq M, Matute C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47:264–272. doi: 10.1016/j.ceca.2009.12.010. - DOI - PubMed
-
- Ando K, Brion JP, Stygelbout V, Suain V, Authelet M, Dedecker R, Chanut A, Lacor P, Lavaur J, Sazdovitch V, et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 2013;125:861–878. doi: 10.1007/s00401-013-1111-z. - DOI - PubMed
-
- Arbel-Ornath M, Hudry E, Boivin JR, Hashimoto T, Takeda S, Kuchibhotla KV, Hou S, Lattarulo CR, Belcher AM, Shakerdge N, et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol Neurodegener. 2017;12:27. doi: 10.1186/s13024-017-0169-9. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
