

Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease
- PMID: 30498206
- PMCID: PMC6265251
- DOI: 10.1038/s41467-018-07466-6
Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease
Abstract
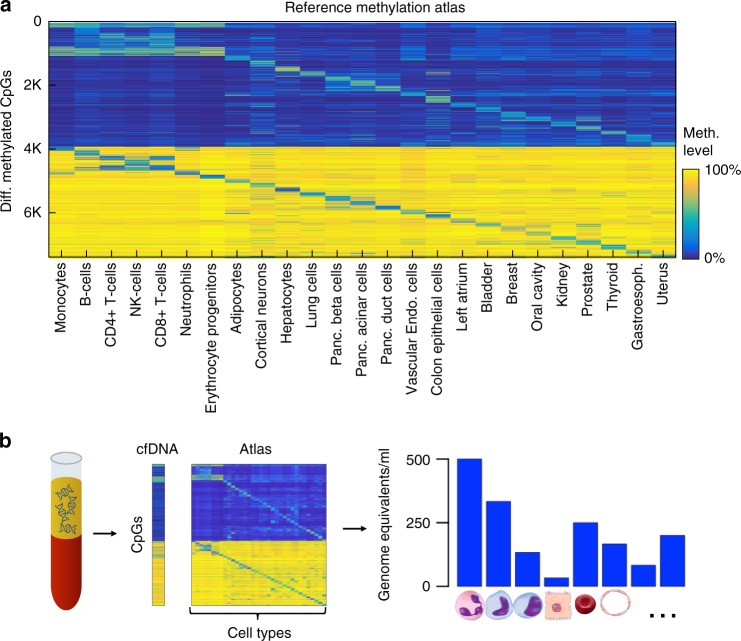
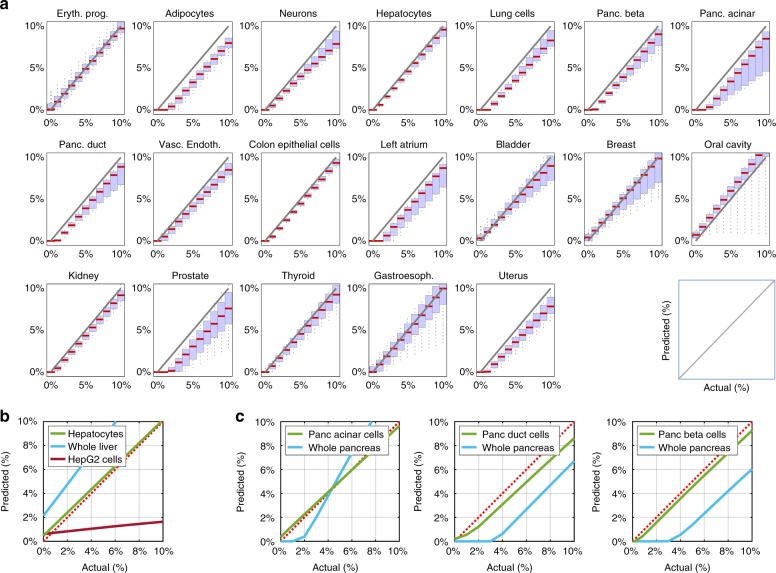
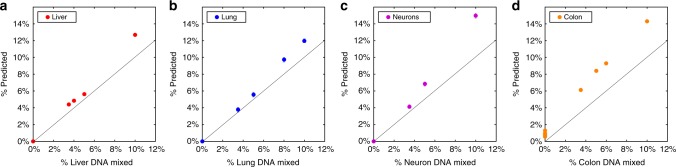
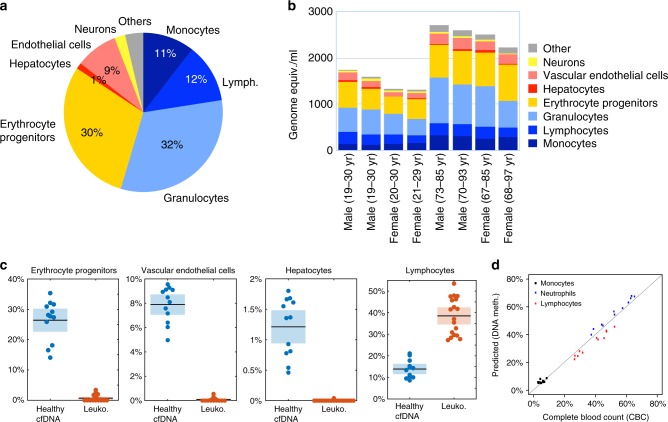
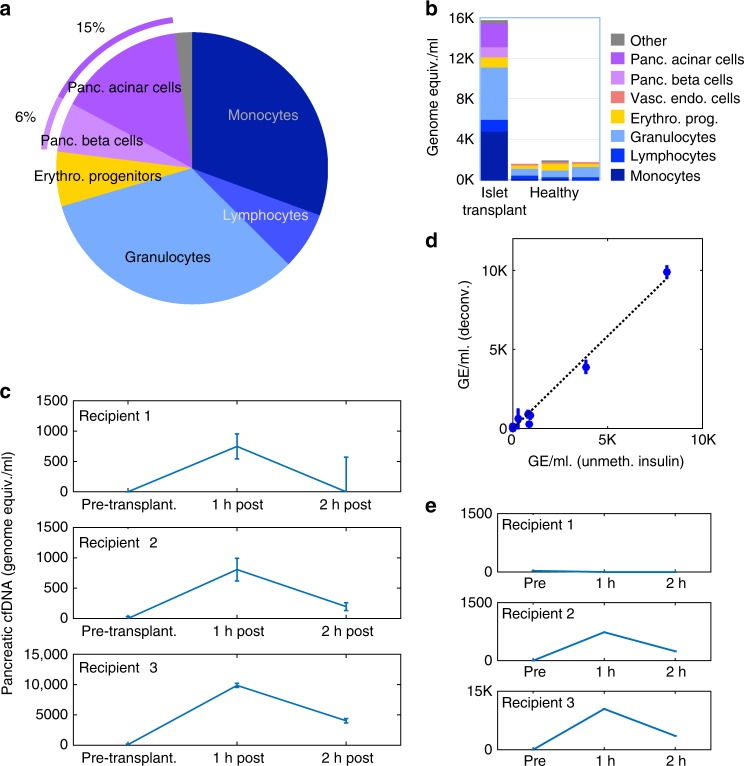
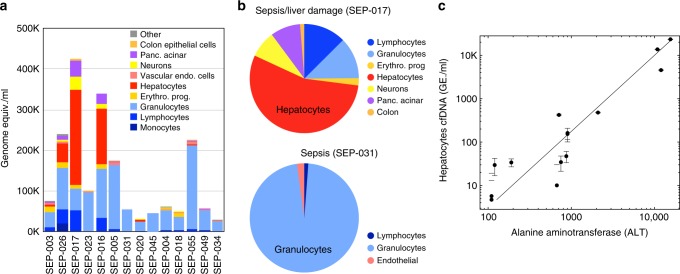
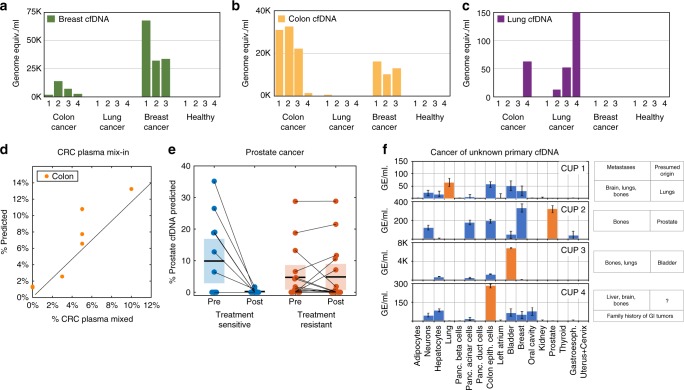
Methylation patterns of circulating cell-free DNA (cfDNA) contain rich information about recent cell death events in the body. Here, we present an approach for unbiased determination of the tissue origins of cfDNA, using a reference methylation atlas of 25 human tissues and cell types. The method is validated using in silico simulations as well as in vitro mixes of DNA from different tissue sources at known proportions. We show that plasma cfDNA of healthy donors originates from white blood cells (55%), erythrocyte progenitors (30%), vascular endothelial cells (10%) and hepatocytes (1%). Deconvolution of cfDNA from patients reveals tissue contributions that agree with clinical findings in sepsis, islet transplantation, cancer of the colon, lung, breast and prostate, and cancer of unknown primary. We propose a procedure which can be easily adapted to study the cellular contributors to cfDNA in many settings, opening a broad window into healthy and pathologic human tissue dynamics.
Conflict of interest statement
J.M., R.S., B.G., T.K. and Y.D. are inventors on a patent entitled “CELL FREE DNA DECONVOLUTION AND USE THEREOF” (US provisional application No. 62/661,179). The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
