

OUTRIDER: A Statistical Method for Detecting Aberrantly Expressed Genes in RNA Sequencing Data
- PMID: 30503520
- PMCID: PMC6288422
- DOI: 10.1016/j.ajhg.2018.10.025
OUTRIDER: A Statistical Method for Detecting Aberrantly Expressed Genes in RNA Sequencing Data
Abstract
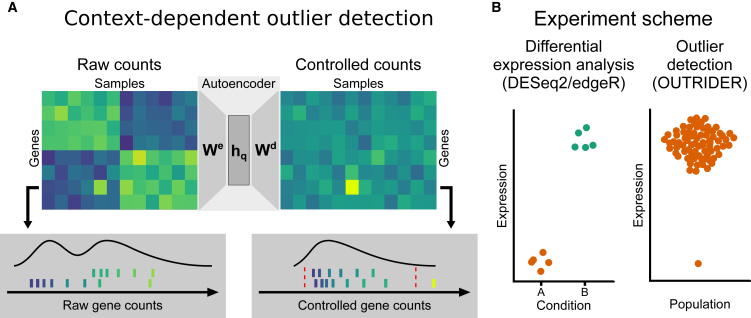
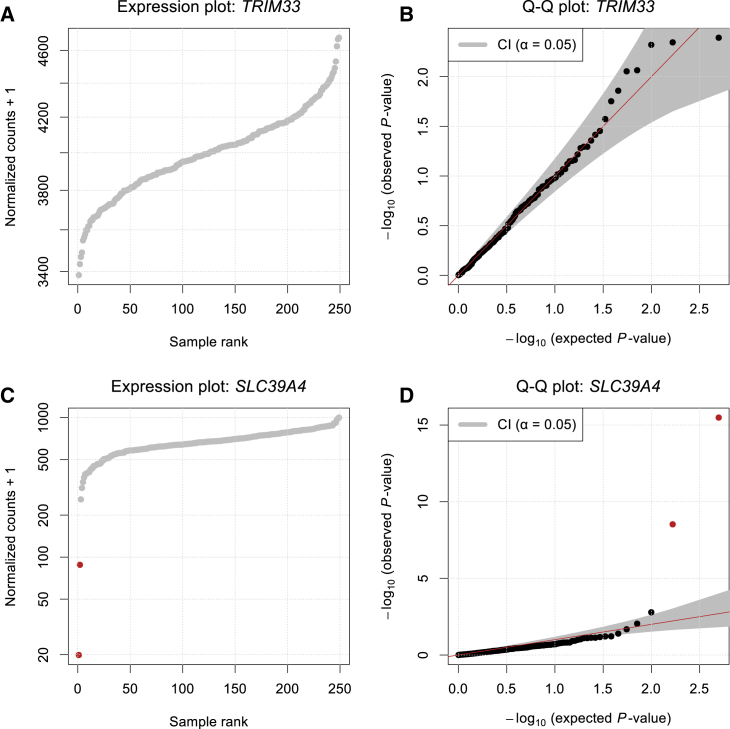
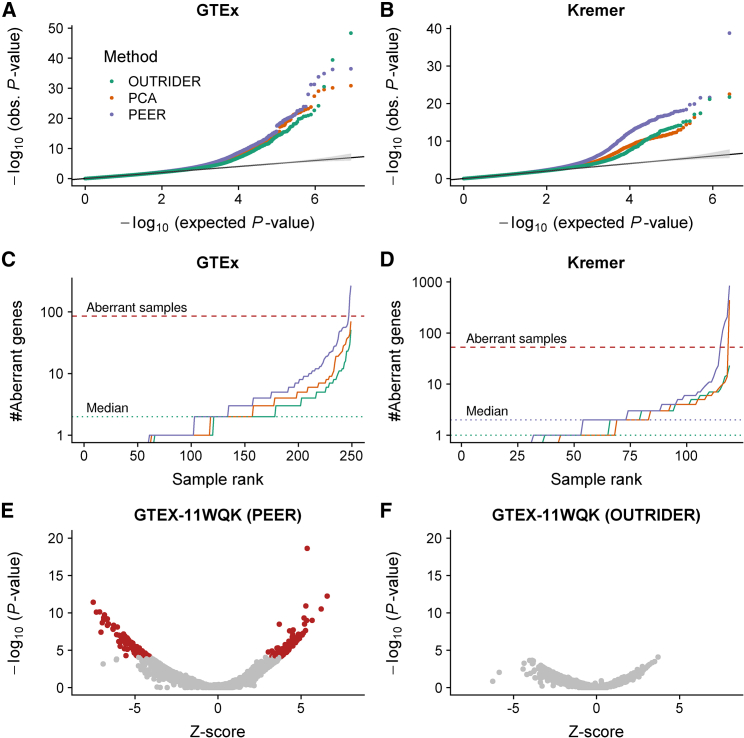
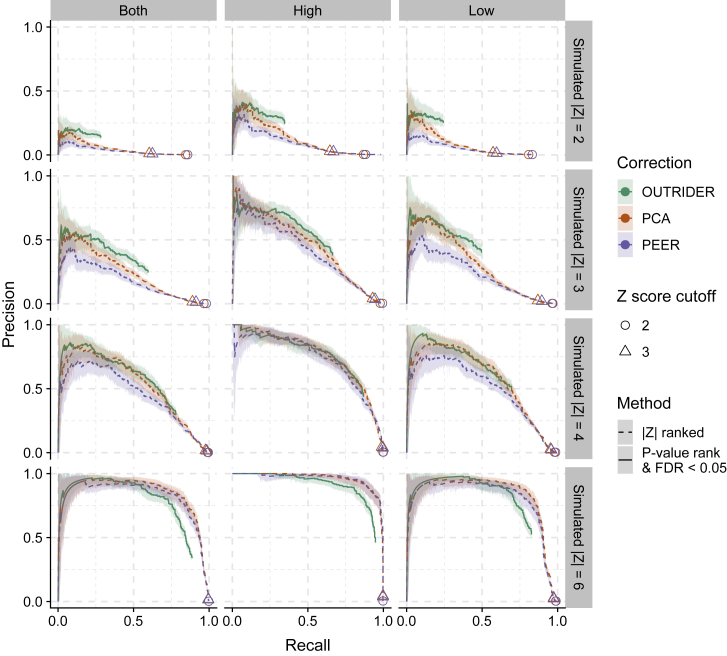
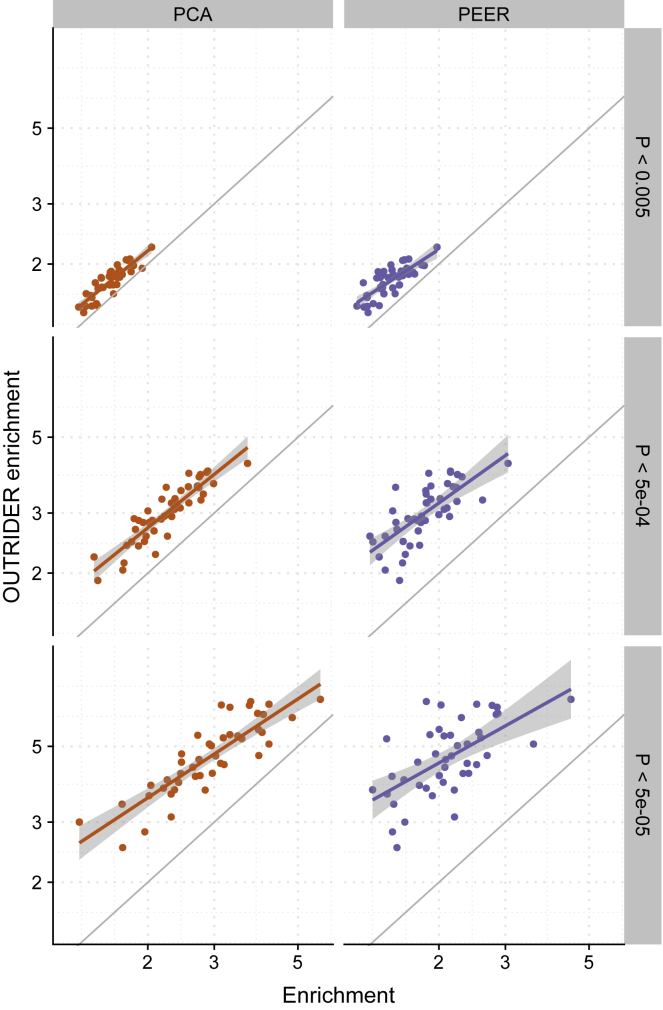
RNA sequencing (RNA-seq) is gaining popularity as a complementary assay to genome sequencing for precisely identifying the molecular causes of rare disorders. A powerful approach is to identify aberrant gene expression levels as potential pathogenic events. However, existing methods for detecting aberrant read counts in RNA-seq data either lack assessments of statistical significance, so that establishing cutoffs is arbitrary, or rely on subjective manual corrections for confounders. Here, we describe OUTRIDER (Outlier in RNA-Seq Finder), an algorithm developed to address these issues. The algorithm uses an autoencoder to model read-count expectations according to the gene covariation resulting from technical, environmental, or common genetic variations. Given these expectations, the RNA-seq read counts are assumed to follow a negative binomial distribution with a gene-specific dispersion. Outliers are then identified as read counts that significantly deviate from this distribution. The model is automatically fitted to achieve the best recall of artificially corrupted data. Precision-recall analyses using simulated outlier read counts demonstrated the importance of controlling for covariation and significance-based thresholds. OUTRIDER is open source and includes functions for filtering out genes not expressed in a dataset, for identifying outlier samples with too many aberrantly expressed genes, and for detecting aberrant gene expression on the basis of false-discovery-rate-adjusted p values. Overall, OUTRIDER provides an end-to-end solution for identifying aberrantly expressed genes and is suitable for use by rare-disease diagnostic platforms.
Keywords: RNA sequencing; aberrant gene expression; normalization; outlier detection; rare disease.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Wright C.F., FitzPatrick D.R., Firth H.V. Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 2018;19:253–268. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
