

doi: 10.1038/s41598-018-35727-3.
A novel cloning strategy for one-step assembly of multiplex CRISPR vectors
Affiliations
- PMID: 30504793
- PMCID: PMC6269432
- DOI: 10.1038/s41598-018-35727-3
Item in Clipboard
A novel cloning strategy for one-step assembly of multiplex CRISPR vectors
Sci Rep.
.
Abstract
One key advantage of the CRISPR/Cas9 system in comparison with other gene editing approaches lies in its potential for multiplexing. Here, we describe an elaborate procedure that allows the assembly of multiple gRNA expression cassettes into a vector of choice within a single step, termed ASAP(Adaptable System for Assembly of multiplexed Plasmids)-cloning. We demonstrate the utility of ASAP-cloning for multiple CRISPR-mediated applications, including efficient multiplex gene editing, robust transcription activation and convenient analysis of Cas9 activity in the presence of multiple gRNAs.
Conflict of interest statement
The authors declare no competing interests.
Figures
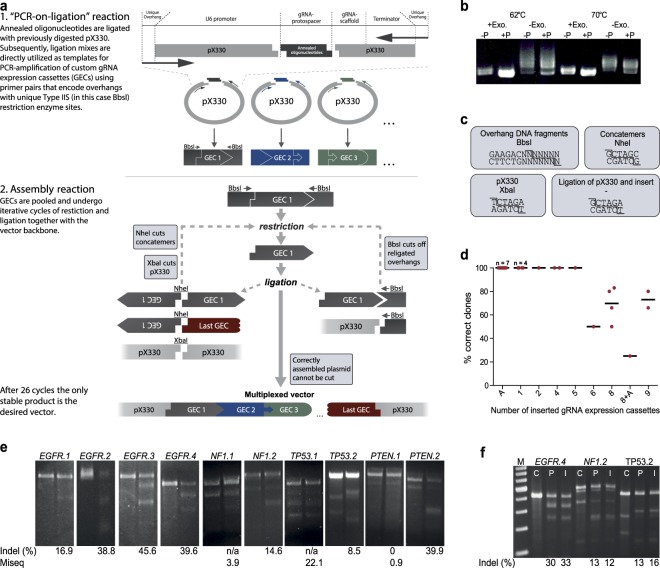
ASAP-cloning allows rapid vector generation for efficient multiplex gene targeting. (a) Workflow of ASAP-cloning. Annealed oligonucleotides, encoding the protospacer complementary region of the gRNA are ligated with the pX330 vector. Subsequently, gRNA expression cassettes (GECs), including the U6 promoter, the gRNA and a transcriptional terminator are amplified directly from the ligation mix. The utilized primers encode for unique restriction enzyme sites that are attached to the GEC during PCR-amplification. In the following assembly reaction, addition of distinct enzymes to cleave GEC overhangs allows to array the respective GECs in a defined order and to insert them into a vector of choice within one process. The only stable plasmid vacant of functional restriction sites during the reaction is the desired product, which is sufficiently enriched after 26 cycles of restriction and ligation. (b) Exonuclease treatment and the use of non-dephosphorylated backbones are crucial for the generation of distinct PCR products. After ligation of annealed oligodinucleotides with either dephosphorylated (−P) or non-dephosphorylated (+P), BbsI-cut pX330 vector, an exonuclease treatment was performed for half of the analyzed ligation mixes. Subsequently, reaction mixes of either the exonuclease treatment (+Exo.) or the ligation (−Exo.) were used as template for a PCR. Annealing temperatures of 62 °C or 70 °C were used, as indicated. (c) Restriction enzyme sites utilized in the construction of pX330-10x. The boxes indicate which DNA molecule was cut with which enzyme and the respective sequence. (d) Efficiency of ASAP-cloning depends on the number of GECs forming the combined insert. Each dot represents an independent cloning approach with the indicated, inserted number of GECs. Bars indicate mean values. Correctness of clones was analyzed via control restriction and Sanger sequencing. “A” indicates the addition of annealed oligonucleotides to the reaction mix. During ASAP-cloning these were inserted into the designated gRNA expression cassette of the original pX330 vector. (e) SURVEYOR assays to determine the frequency of generated indels at targeted loci. Each locus was amplified from control gDNA (left lane) and from gDNA isolated from cells that had been transfected with pX330-10x (right lane). Resulting PCR products were used in SURVEYOR assays. For some loci, control DNA was digested as well, indicating PCR artifacts or endogenous SNVs. In this case, the resulting bands were added to the “undigested” fraction when calculating indel frequencies. Loci for which the SURVEYOR assay did not yield measurable results were sequenced on the MiSeq platform. (f) pX330-10x and pX330-impsc-10x display similar genome editing efficiencies. SURVEYOR assays of the indicated PCR products were performed after isolation of gDNA from untransfected cells (C) or cells that had been transfected with either pX330-10x (P) or pX330-impsc-10x (I). M = GeneRuler 100 bp DNA Ladder.
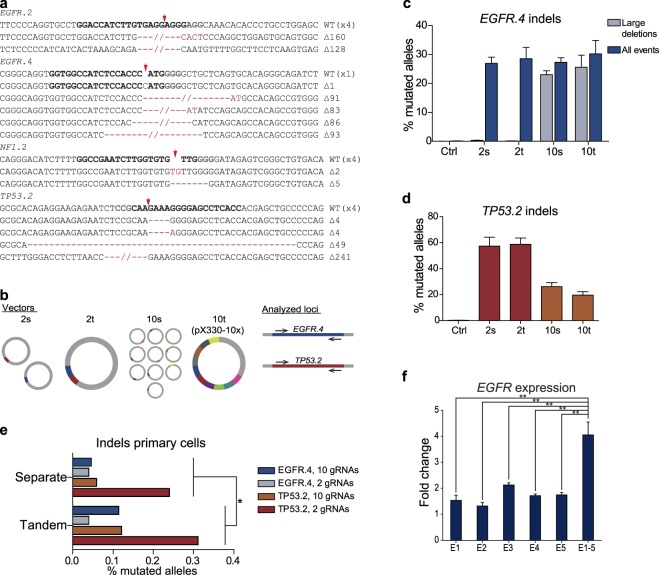
Multiplexed vectors constructed by ASAP cloning outperform single plasmids. (a) Sanger sequencing of indicated loci after transfection of HEK293T cells with pX330-10x displays high amount of alleles with frameshifting indels. (b) Illustration of the utilized vectors and the analyzed loci of the following experiments. 2 s = Two separate plasmids encoding gRNAs which target the EGFR.4 and the TP53.2 locus, respectively; 2t = single vector encoding these two gRNA cassettes in tandem; 10 s = Ten separate plasmids with ten different genomic targets including the analyzed loci; 10t = One single tandem vector (pX330-10x) encoding gRNAs to target these ten loci. (c,d) Targeted deep sequencing of the indicated loci after transfection of HEK293T cells with the aforementioned vectors to identify genomic editing events. Deletions encompassing two of the target sites at the EGFR locus being 85 bp apart (EGFR.3 and EGFR.4) were counted and declared as large deletions. Ctrl = untransfected cells; n = 3 (biological replicates). Data are displayed as mean + standard deviation. (e) Allele editing events after transfection of primary glioblastoma cells with the indicated, aforementioned vectors. To account for artificial indels at the analyzed loci, respective mutated allele frequencies of WT cells (0.0144% for TP53.2 and 0.07655% for EGFR.4) were subtracted from mutated allele frequencies of samples. The analyzed loci and the number of delivered gRNAs are depicted in the figure legend. Results of approaches in which separate vectors have been transfected were plotted against tandem approaches. n = 1. P-value ≤ 0.029. The P-value was calculated using a one-sided, paired t-test. (f) HEK293T cells were transfected with pcDNA-dCas9-VP64 vectors in which either one separate gRNA expression cassette (E1, E2, E3, E4 or E5) or all together (E1-5) had been integrated via ASAP-cloning. The respective gRNAs target the promoter region of EGFR. Total RNA was isolated and expression of EGFR was assessed by real-time PCR. The graph indicates the fold change of expression compared to cells that had been transfected with an empty vector. P-values: E1 vs. E1-5 ≤ 0.0017, E2 vs. E1-5 ≤ 0.0014, E3 vs. E1-5 ≤ 0.0086, E4 vs. E1-5 ≤ 0.0039, E5 vs. E1-5 ≤ 0.0043. n = 3 (biological replicates). The data are displayed as mean + SEM.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
