

High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries
- PMID: 30504855
- PMCID: PMC6269478
- DOI: 10.1038/s41467-018-07641-9
High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries
Abstract
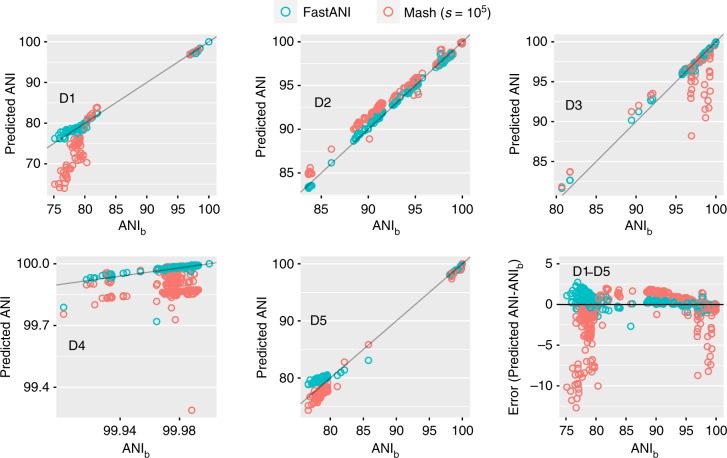
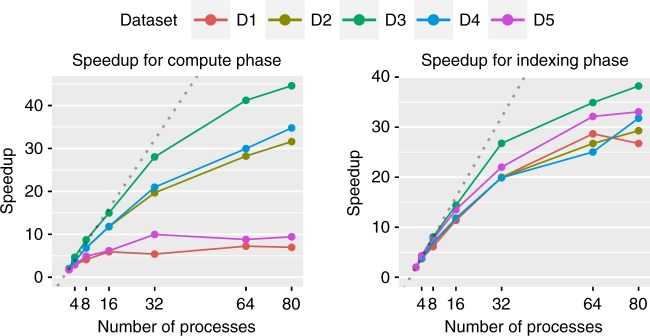
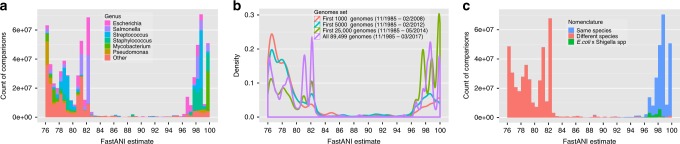
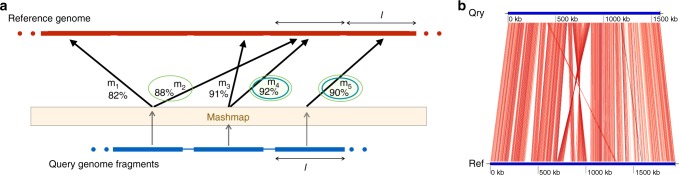
A fundamental question in microbiology is whether there is continuum of genetic diversity among genomes, or clear species boundaries prevail instead. Whole-genome similarity metrics such as Average Nucleotide Identity (ANI) help address this question by facilitating high resolution taxonomic analysis of thousands of genomes from diverse phylogenetic lineages. To scale to available genomes and beyond, we present FastANI, a new method to estimate ANI using alignment-free approximate sequence mapping. FastANI is accurate for both finished and draft genomes, and is up to three orders of magnitude faster compared to alignment-based approaches. We leverage FastANI to compute pairwise ANI values among all prokaryotic genomes available in the NCBI database. Our results reveal clear genetic discontinuity, with 99.8% of the total 8 billion genome pairs analyzed conforming to >95% intra-species and <83% inter-species ANI values. This discontinuity is manifested with or without the most frequently sequenced species, and is robust to historic additions in the genome databases.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Systematics: The Cohesive Nature of Bacterial Species Taxa.Curr Biol. 2019 Mar 4;29(5):R169-R172. doi: 10.1016/j.cub.2019.01.033. Curr Biol. 2019. PMID: 30836089
-
Reply to: "Re-evaluating the evidence for a universal genetic boundary among microbial species".Nat Commun. 2021 Jul 7;12(1):4060. doi: 10.1038/s41467-021-24129-1. Nat Commun. 2021. PMID: 34234115 Free PMC article. No abstract available.
-
Re-evaluating the evidence for a universal genetic boundary among microbial species.Nat Commun. 2021 Jul 7;12(1):4059. doi: 10.1038/s41467-021-24128-2. Nat Commun. 2021. PMID: 34234129 Free PMC article. No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
