

Deep generative modeling for single-cell transcriptomics
- PMID: 30504886
- PMCID: PMC6289068
- DOI: 10.1038/s41592-018-0229-2
Deep generative modeling for single-cell transcriptomics
Abstract
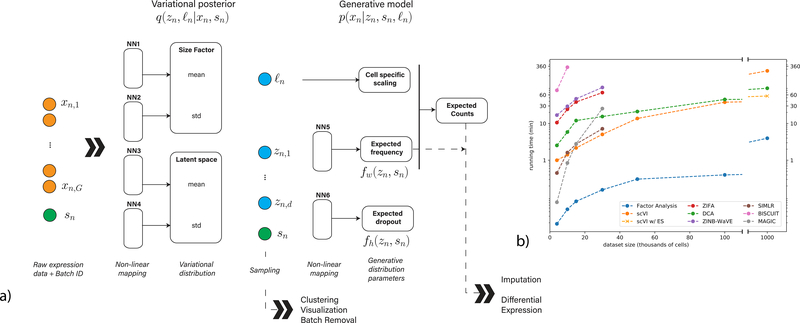
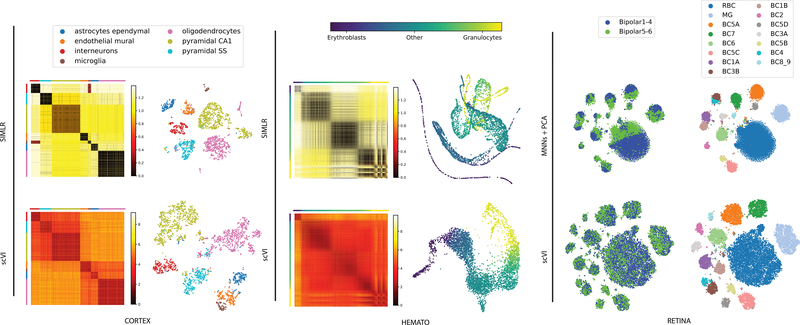
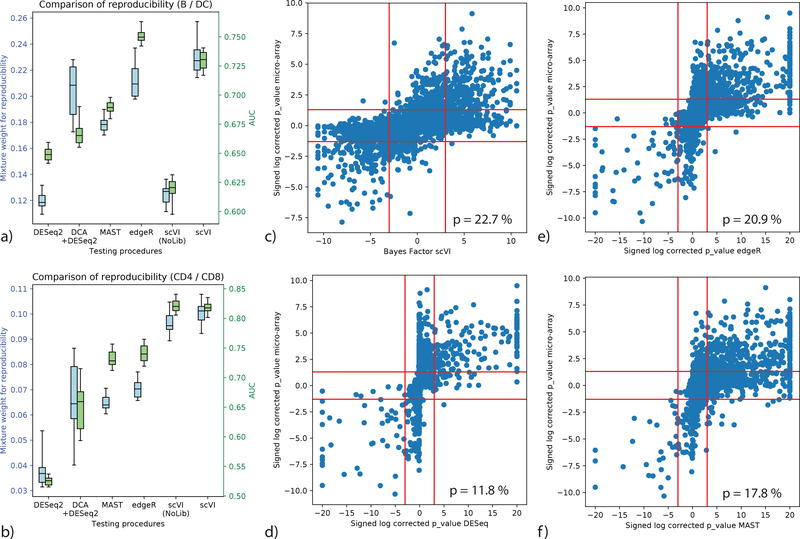
Single-cell transcriptome measurements can reveal unexplored biological diversity, but they suffer from technical noise and bias that must be modeled to account for the resulting uncertainty in downstream analyses. Here we introduce single-cell variational inference (scVI), a ready-to-use scalable framework for the probabilistic representation and analysis of gene expression in single cells ( https://github.com/YosefLab/scVI ). scVI uses stochastic optimization and deep neural networks to aggregate information across similar cells and genes and to approximate the distributions that underlie observed expression values, while accounting for batch effects and limited sensitivity. We used scVI for a range of fundamental analysis tasks including batch correction, visualization, clustering, and differential expression, and achieved high accuracy for each task.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Comment in
-
Bayesian deep learning for single-cell analysis.Nat Methods. 2018 Dec;15(12):1009-1010. doi: 10.1038/s41592-018-0230-9. Nat Methods. 2018. PMID: 30504887 No abstract available.
