

iHam and pyHam: visualizing and processing hierarchical orthologous groups
- PMID: 30508066
- PMCID: PMC6612847
- DOI: 10.1093/bioinformatics/bty994
iHam and pyHam: visualizing and processing hierarchical orthologous groups
Abstract
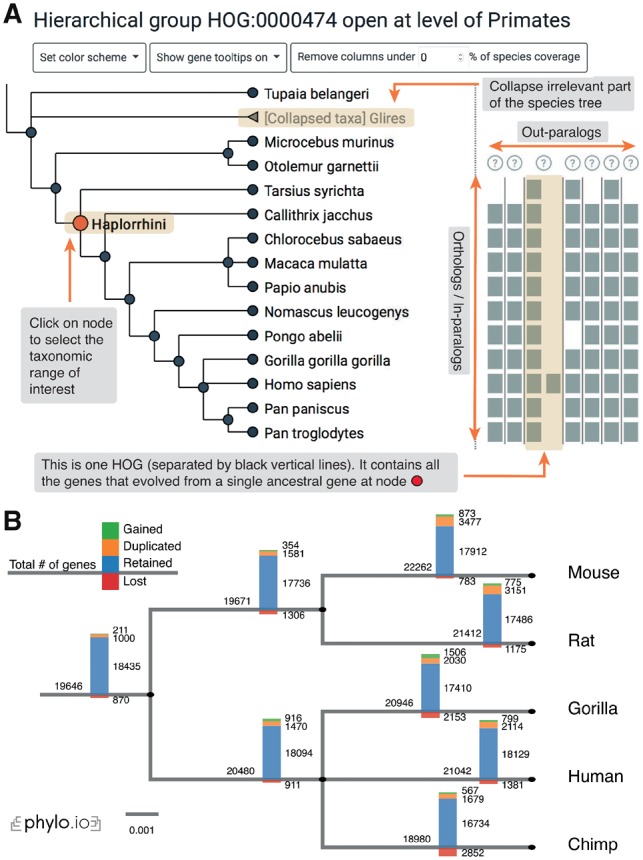
Summary: The evolutionary history of gene families can be complex due to duplications and losses. This complexity is compounded by the large number of genomes simultaneously considered in contemporary comparative genomic analyses. As provided by several orthology databases, hierarchical orthologous groups (HOGs) are sets of genes that are inferred to have descended from a common ancestral gene within a species clade. This implies that the set of HOGs defined for a particular clade correspond to the ancestral genes found in its last common ancestor. Furthermore, by keeping track of HOG composition along the species tree, it is possible to infer the emergence, duplications and losses of genes within a gene family of interest. However, the lack of tools to manipulate and analyse HOGs has made it difficult to extract, display and interpret this type of information. To address this, we introduce interactive HOG analysis method, an interactive JavaScript widget to visualize and explore gene family history encoded in HOGs and python HOG analysis method, a python library for programmatic processing of genes families. These complementary open source tools greatly ease adoption of HOGs as a scalable and interpretable concept to relate genes across multiple species.
Availability and implementation: iHam's code is available at https://github.com/DessimozLab/iHam or can be loaded dynamically. pyHam's code is available at https://github.com/DessimozLab/pyHam and or via the pip package 'pyham'.
© The Author(s) 2018. Published by Oxford University Press.
Figures
References
-
- Chevenet F. et al. (2016) Berry SylvX: a viewer for phylogenetic tree reconciliations. Bioinformatics, 32, 608–610. - PubMed
-
- Dufayard J.-F. et al. (2005) Tree pattern matching in phylogenetic trees: automatic search for orthologs or paralogs in homologous gene sequence databases. Bioinformatics, 21, 2596–2603. - PubMed
-
- Fitch W.M. (1970) Distinguishing homologous from analogous proteins. Syst. Zool., 19, 99–113. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
