

Mutant p53 in cancer therapy-the barrier or the path
- PMID: 30508182
- PMCID: PMC6487791
- DOI: 10.1093/jmcb/mjy072
Mutant p53 in cancer therapy-the barrier or the path
Abstract
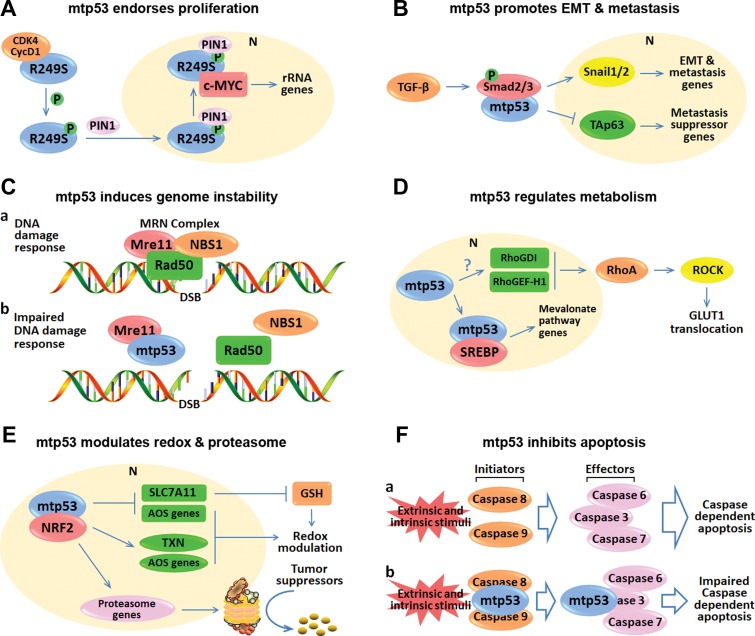
Since wild-type p53 is central for maintaining genomic stability and preventing oncogenesis, its coding gene TP53 is highly mutated in ~50% of human cancers, and its activity is almost abrogated in the rest of cancers. Approximately 80% of p53 mutations are single point mutations with several hotspot mutations. Besides loss of function and dominant-negative effect on the wild-type p53 activity, the hotspot p53 mutants also acquire new oncogenic functions, so-called 'gain-of-functions' (GOF). Because the GOF of mutant p53 is highly associated with late-stage malignance and drug resistance, these p53 mutants have become hot targets for developing novel cancer therapies. In this essay, we review some recent progresses in better understanding of the role of mutant p53 GOF in chemoresistance and the underlying mechanisms, and discuss the pros and cons of targeting mutant p53 for the development of anti-cancer therapies.
Keywords: cancer therapy; chemoresistance; gain-of-function; mutant p53; synthetic lethality.
© The Author(s) (2019). Published by Oxford University Press on behalf of Journal of Molecular Cell Biology, IBCB, SIBS, CAS.
Figures


References
-
- Aas T., Borresen A.L., Geisler S., et al. (1996). Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 2, 811–814. - PubMed
-
- Adorno M., Cordenonsi M., Montagner M., et al. (2009). A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137, 87–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
