

Genomic sequencing is required for identification of tuberculosis transmission in Hawaii
- PMID: 30509214
- PMCID: PMC6276198
- DOI: 10.1186/s12879-018-3502-1
Genomic sequencing is required for identification of tuberculosis transmission in Hawaii
Abstract
Background: Tuberculosis (TB) caused an estimated 1.4 million deaths and 10.4 million new cases globally in 2015. TB rates in the United States continue to steadily decline, yet rates in the State of Hawaii are perennially among the highest in the nation due to a continuous influx of immigrants from the Western Pacific and Asia. TB in Hawaii is composed of a unique distribution of genetic lineages, with the Beijing and Manila families of Mycobacterium tuberculosis (Mtb) comprising over two-thirds of TB cases. Standard fingerprinting methods (spoligotyping plus 24-loci Mycobacterial Interspersed Repetitive Units-Variable Number Tandem Repeats [MIRU-VNTR] fingerprinting) perform poorly when used to identify actual transmission clusters composed of isolates from these two families. Those typing methods typically group isolates from these families into large clusters of non-linked isolates with identical fingerprints. Next-generation whole-genome sequencing (WGS) provides a new tool for molecular epidemiology that can resolve clusters of isolates with identical spoligotyping and MIRU-VNTR fingerprints.
Methods: We performed WGS and SNP analysis and evaluated epidemiological data to investigate 19 apparent TB transmission clusters in Hawaii from 2003 to 2017 in order to assess WGS' ability to resolve putative Mtb clusters from the Beijing and Manila families. This project additionally investigated MIRU-VNTR allele prevalence to determine why standard Mtb fingerprinting fails to usefully distinguish actual transmission clusters from these two Mtb families.
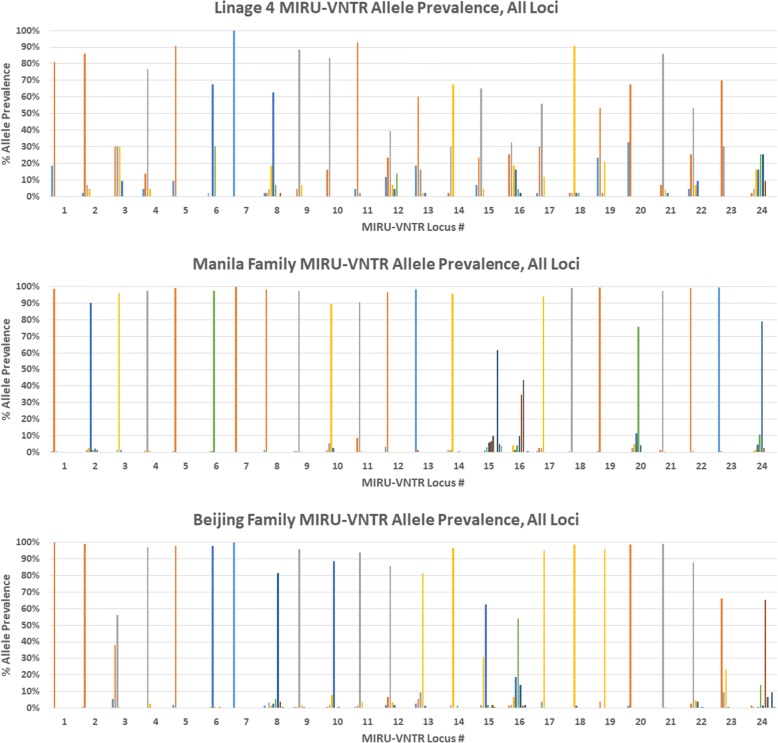
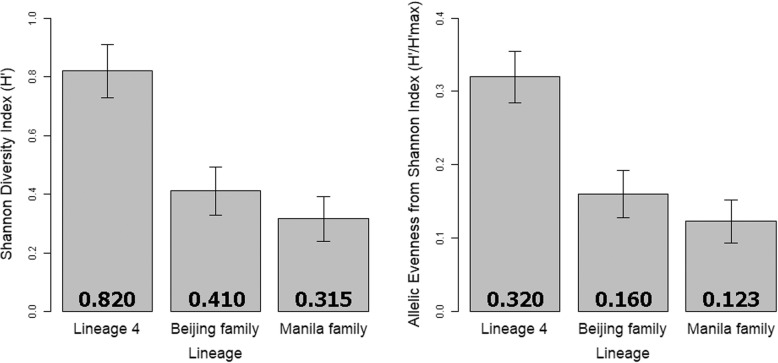
Results: WGS excluded transmission events in seven of these putative clusters, confirmed transmission in eight, and identified both transmission-linked and non-linked isolates in four. For epidemiologically identified clusters, while the sensitivity of MIRU-VNTR fingerprinting for identifying actual transmission clusters was found to be 100%, its specificity was only 28.6% relative to WGS. We identified that the Beijing and Manila families' significantly lower Shannon evenness of MIRU-VNTR allele distributions than lineage 4 was the cause of standard fingerprinting's poor performance when identifying transmission in Beijing and Manila family clusters.
Conclusions: This study demonstrated that WGS is necessary for epidemiological investigation of TB in Hawaii and the Pacific.
Keywords: Genomic epidemiology; Hawaii; Mycobacterium tuberculosis; Spoligotyping.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable – all details relating to patients were de-identified pursuant to State of Hawaii HIPAA guidelines prior to inclusion in this study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Global Tuberculosis Report 2016. http://apps.who.int/medicinedocs/documents/s23098en/s23098en.pdf. Accessed 5 Dec 2017.
-
- Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 2018;16(4):202–13. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
