

The genetic basis of disease
- PMID: 30509934
- PMCID: PMC6279436
- DOI: 10.1042/EBC20170053
The genetic basis of disease
Erratum in
-
Correction: The genetic basis of disease.Essays Biochem. 2020 Oct 8;64(4):681. doi: 10.1042/EBC20170053_COR. Essays Biochem. 2020. PMID: 32720679 Free PMC article. No abstract available.
Abstract
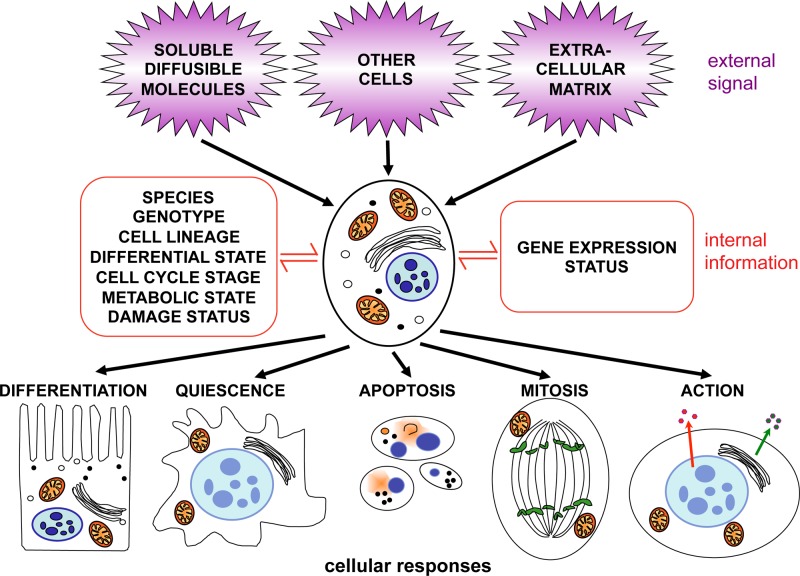
Genetics plays a role, to a greater or lesser extent, in all diseases. Variations in our DNA and differences in how that DNA functions (alone or in combinations), alongside the environment (which encompasses lifestyle), contribute to disease processes. This review explores the genetic basis of human disease, including single gene disorders, chromosomal imbalances, epigenetics, cancer and complex disorders, and considers how our understanding and technological advances can be applied to provision of appropriate diagnosis, management and therapy for patients.
Keywords: cancer; genetics; genomics; molecular basis of health and disease.
© 2018 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures

References
Further reading: The human genome and variation
-
- Murphy E. (2018) Forensic DNA typing. Annu. Rev. Criminol. 1, 497–515 10.1146/annurev-criminol-032317-092127 - DOI
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J.. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 10.1038/gim.2015.30 - DOI - PMC - PubMed
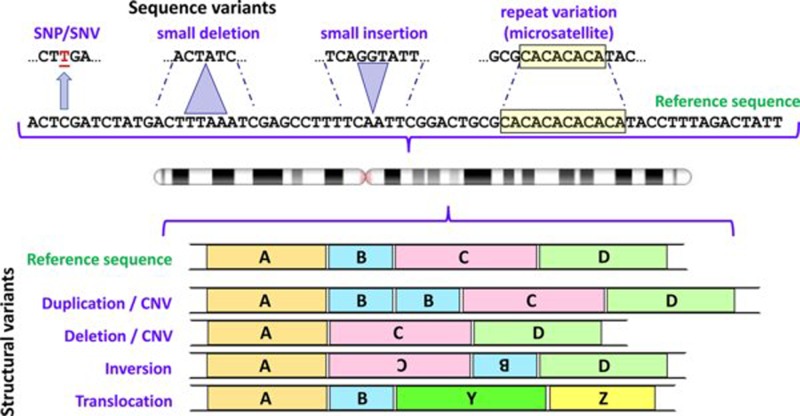
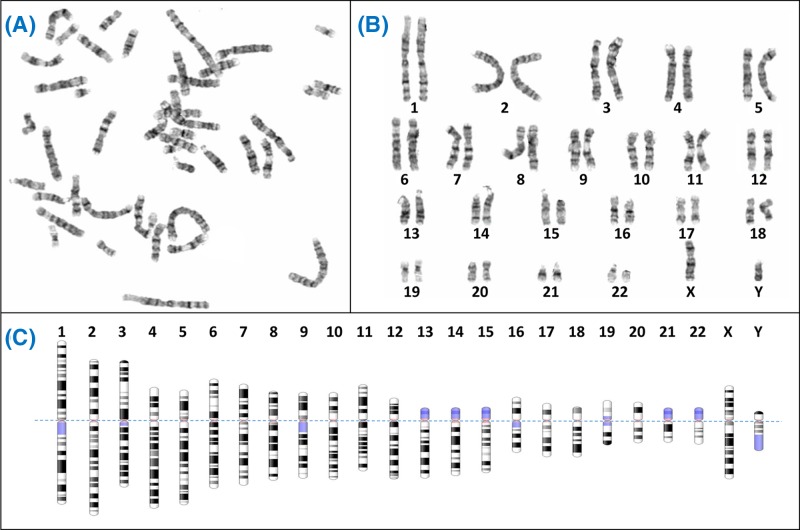
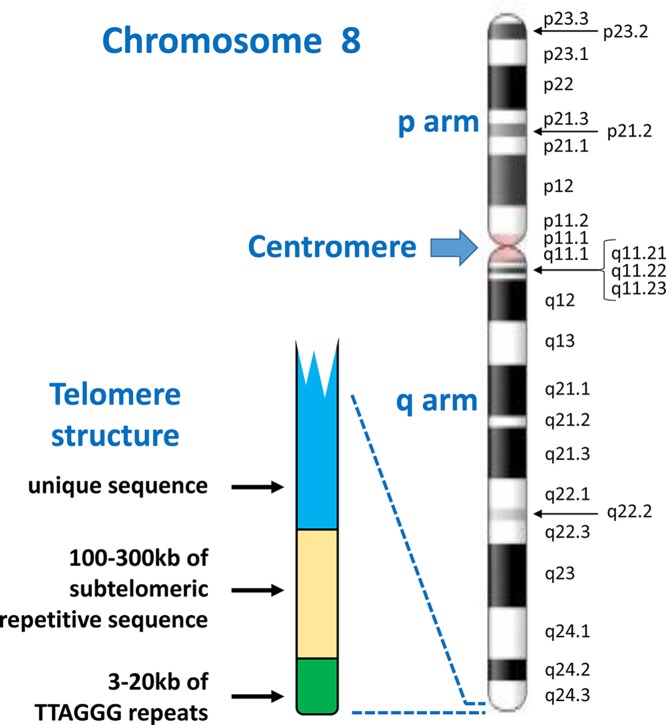
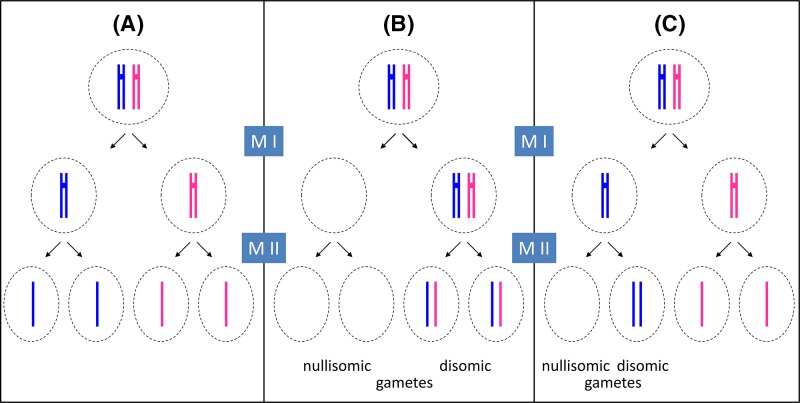
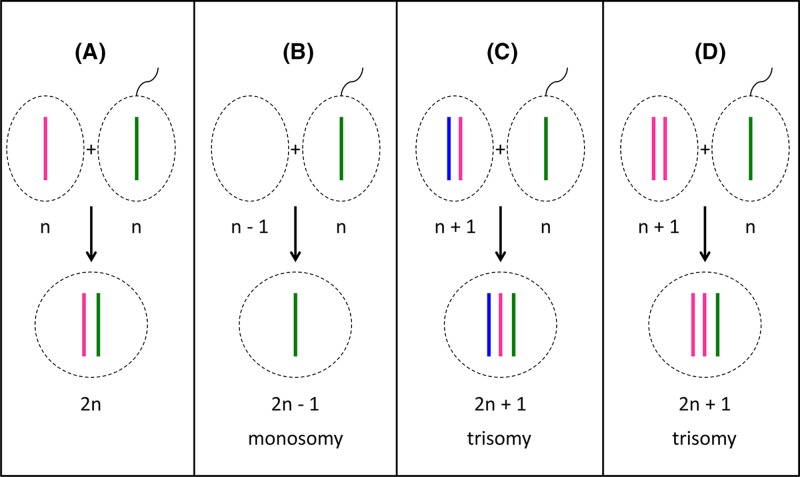
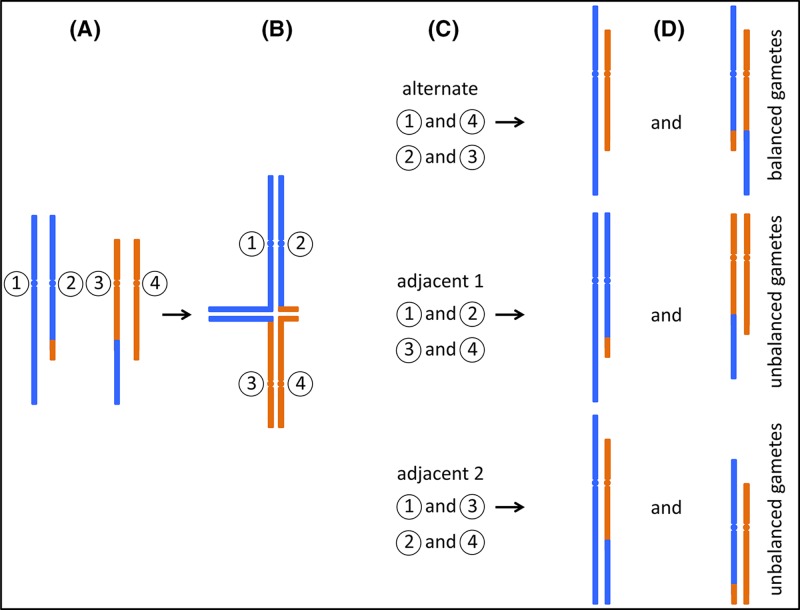
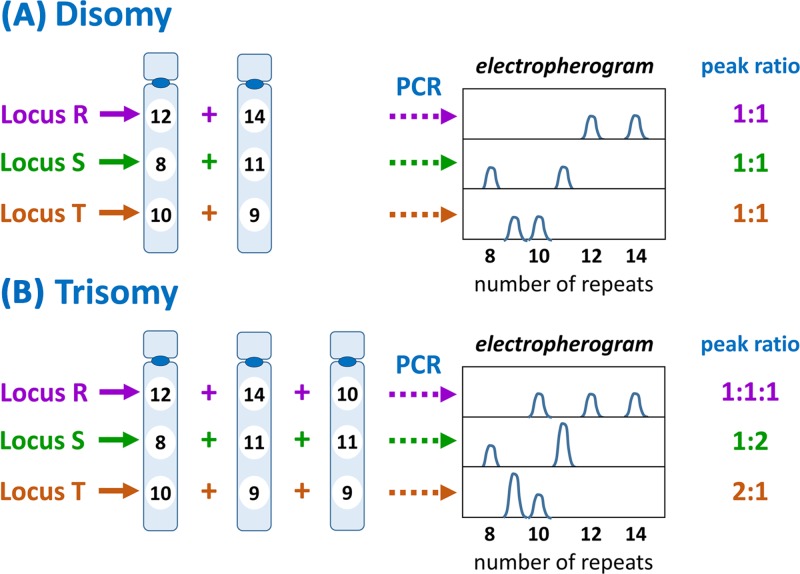
Chromosome structure and chromosomal disorders
-
- To learn more about general genetics, consult one of many available text books. The following is freely available online.
-
- Griffiths A.J.F, Miller J.H., Suzuki D.T., Lewontin R.C. and Gelbart W.M. (2000) An Introduction to Genetic Analysis, 7th edn, W.H. Freeman, New York, https://www.ncbi.nlm.nih.gov/books/NBK21766/
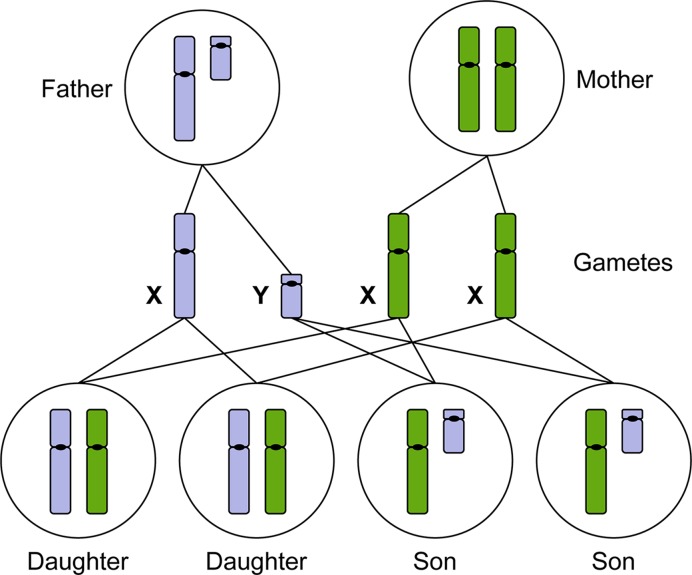
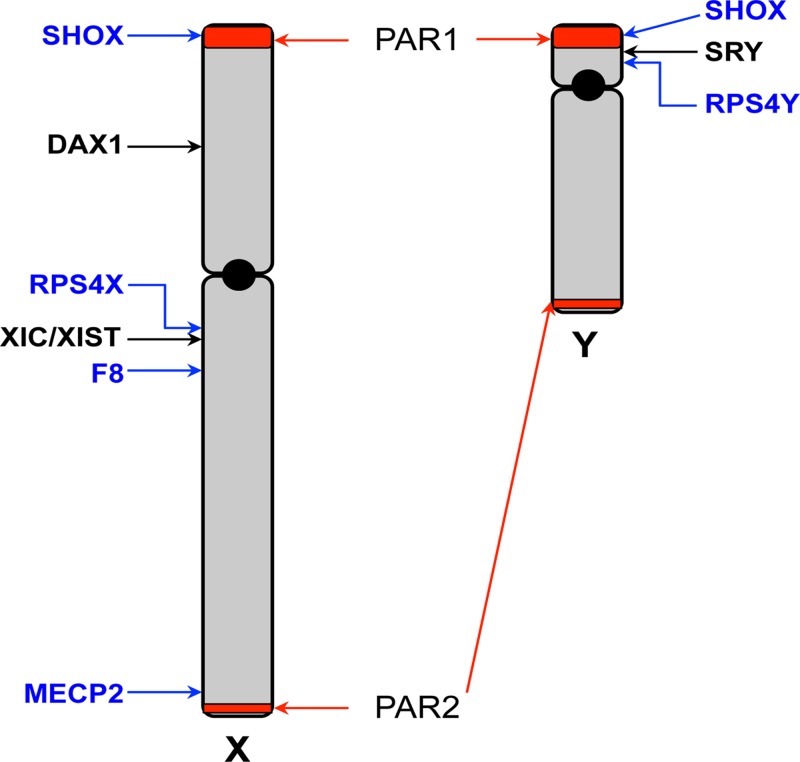
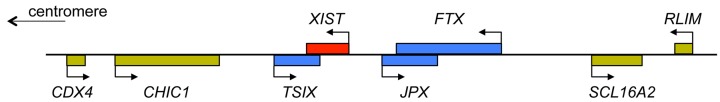
The sex chromosomes, X and Y
-
- Bonora G. and Disteche C.M. (2017) Structural aspects of the inactive X chromosome. Phil. Trans. R. Soc. B Biol. Sci. 372, 20160357, (and others in this volume of 12 contributions to a discussion meeting issue ‘X-chromsome inactivation: a tribute to Mary Lyon’) 10.1098/rstb.2016.0357 - DOI - PMC - PubMed
-
- Gilbert S.F. (2000) Developmental Biology, 6th edn, Chromosomal Sex Determination in Mammals Sinauer Associates
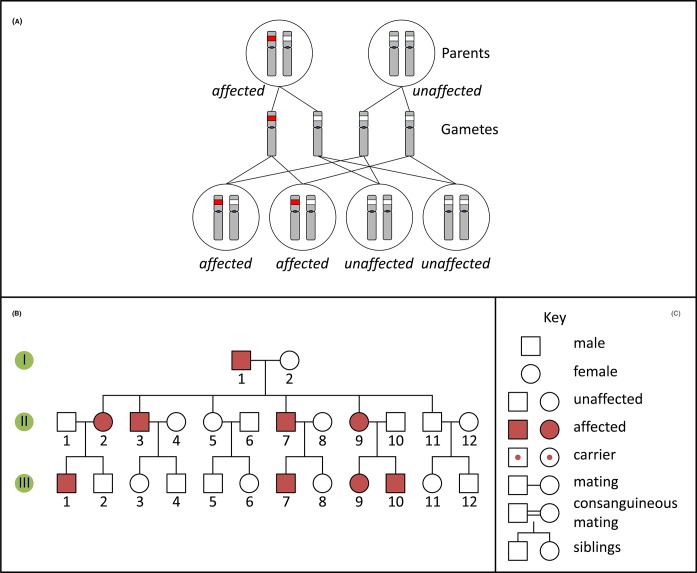
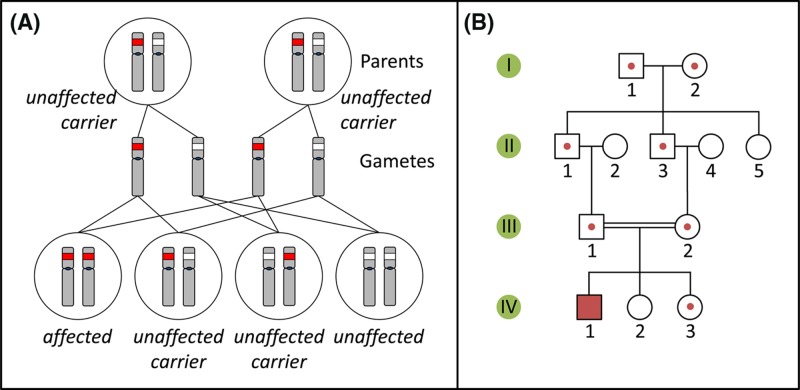
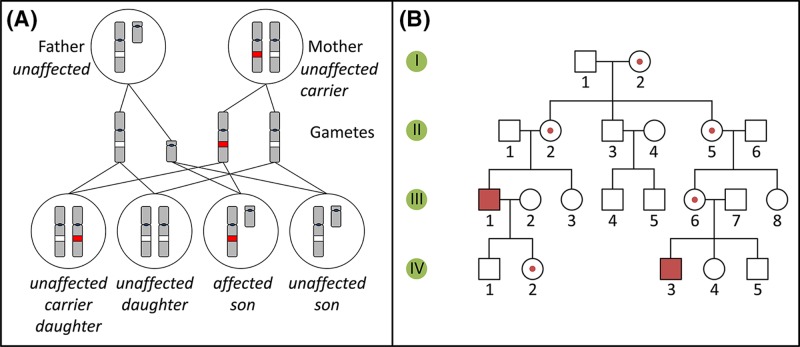
Single-gene disorders
-
- Chial H. (2008) Mendelian genetics: patterns of inheritance and single-gene disorders. Nat. Education 1, 63
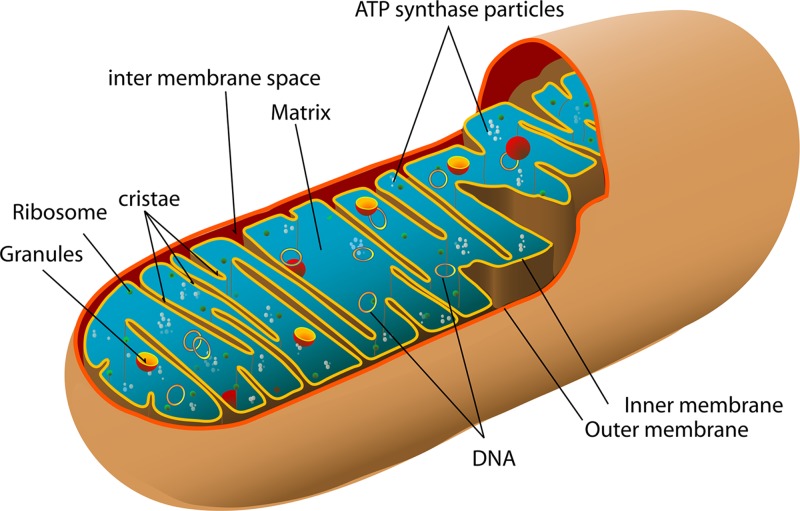
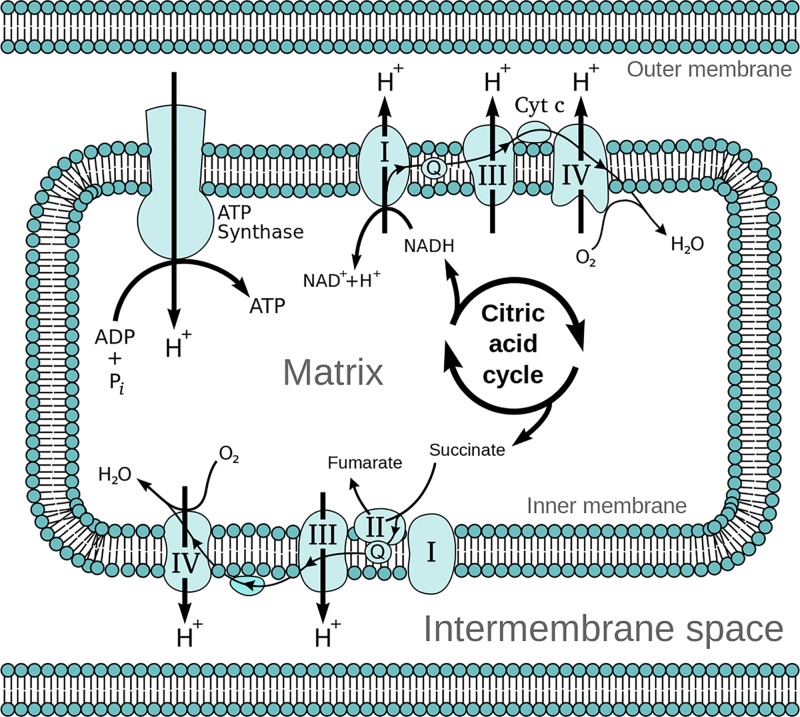
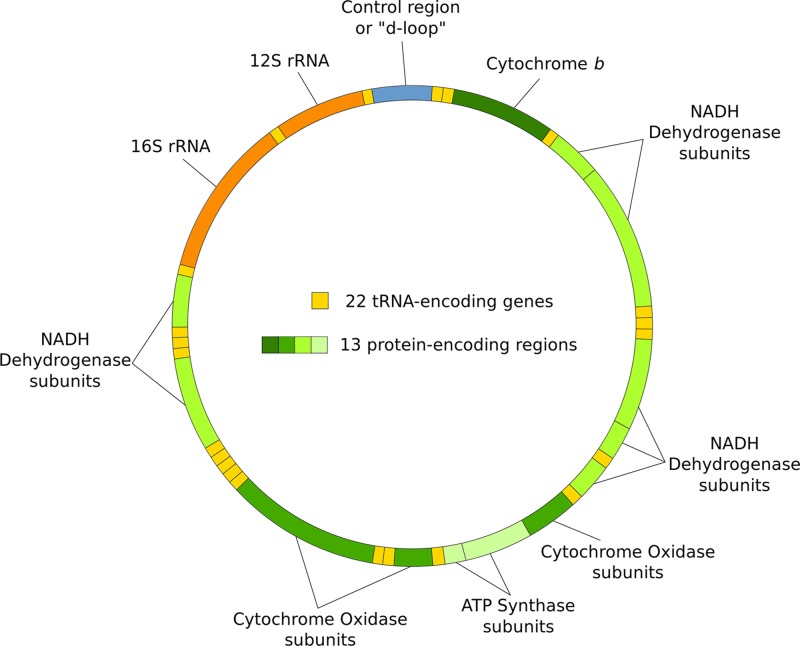
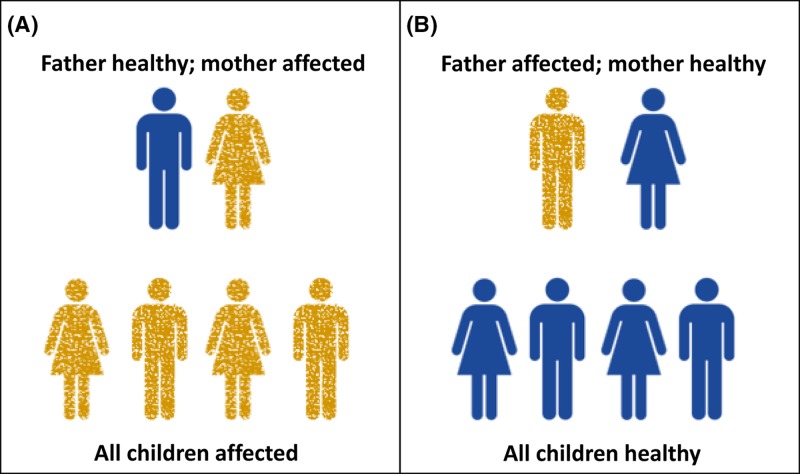
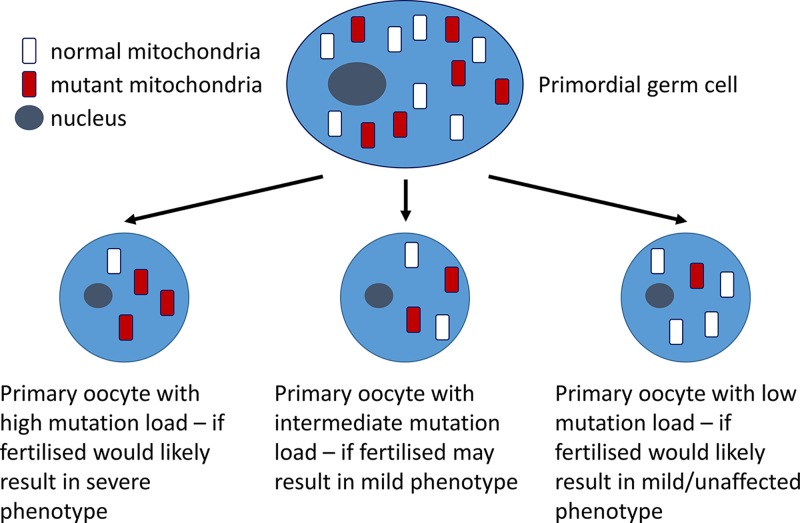
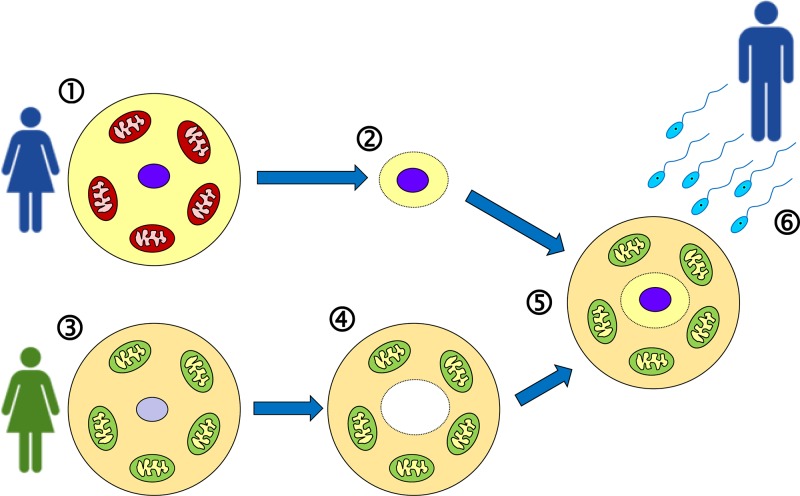
Mitochondrial disorders
-
- Chinnery P. (2000) Mitochondrial Disorders Overview. SourceGeneReviews® (Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K. and Amemiya A., eds), pp. 1993–2018, University of Washington, Seattle, Seattle (WA)
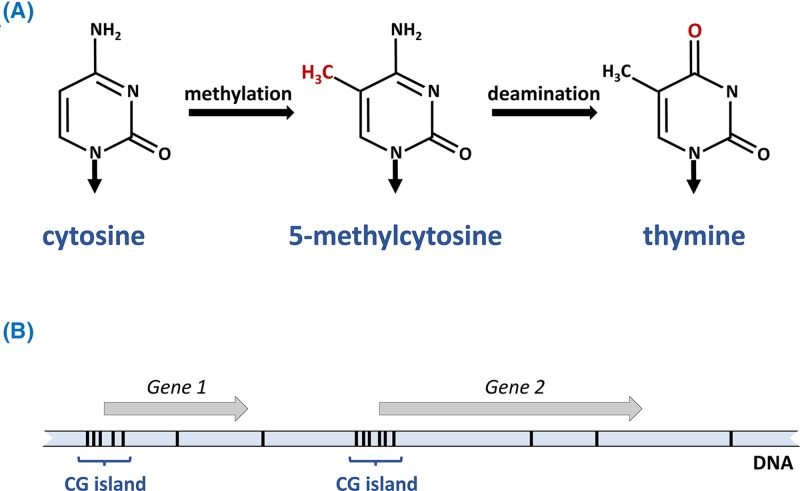
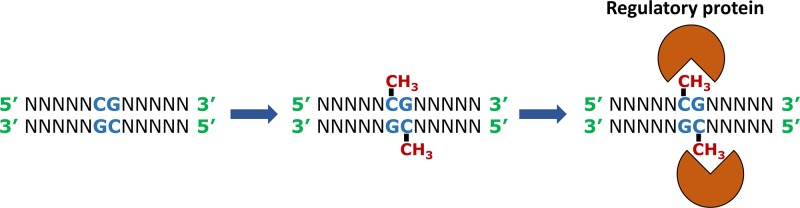
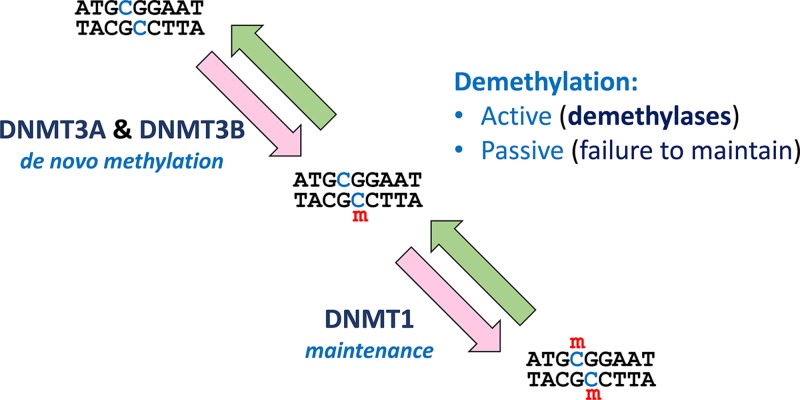
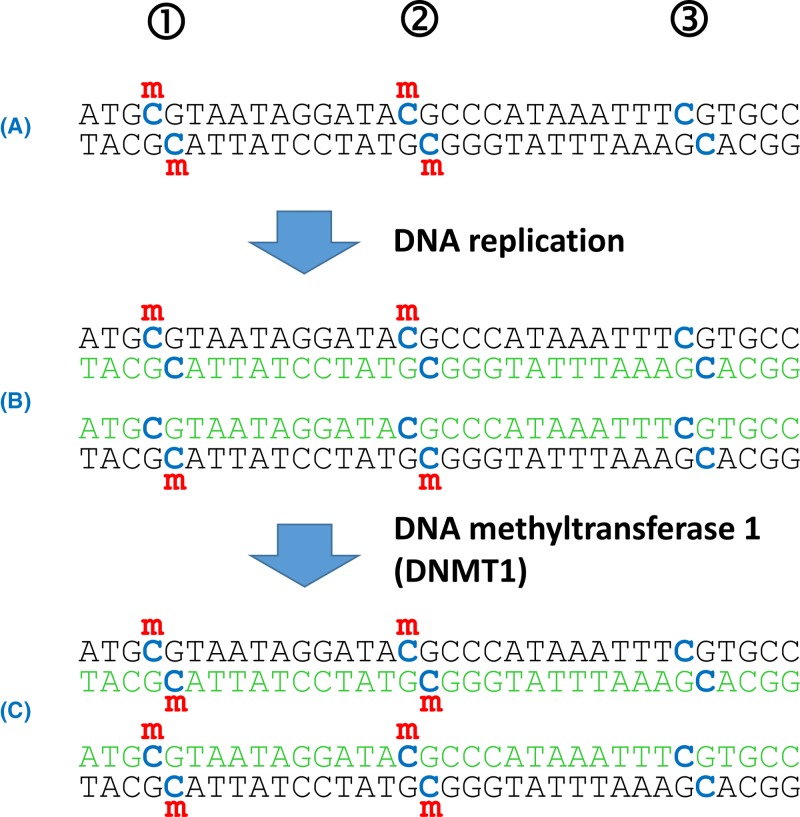
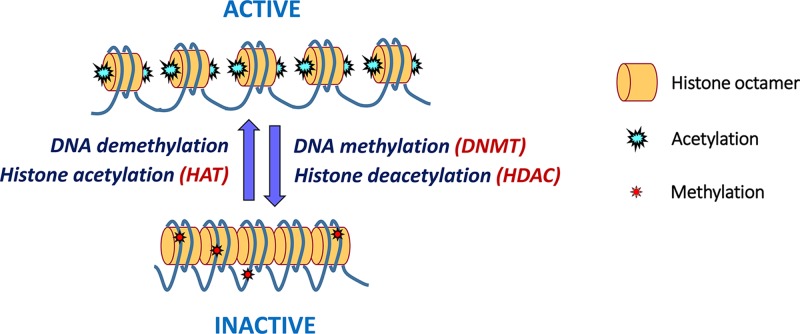
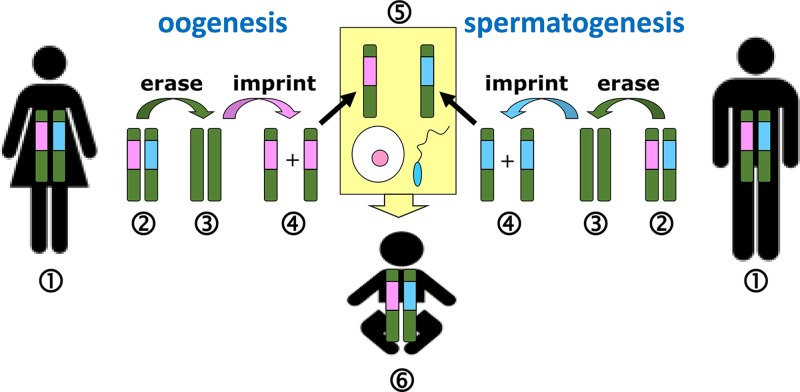
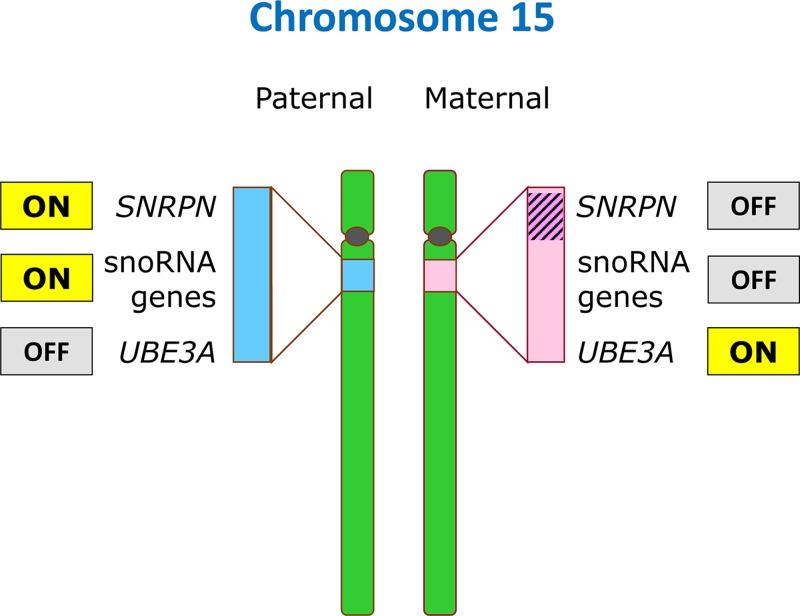
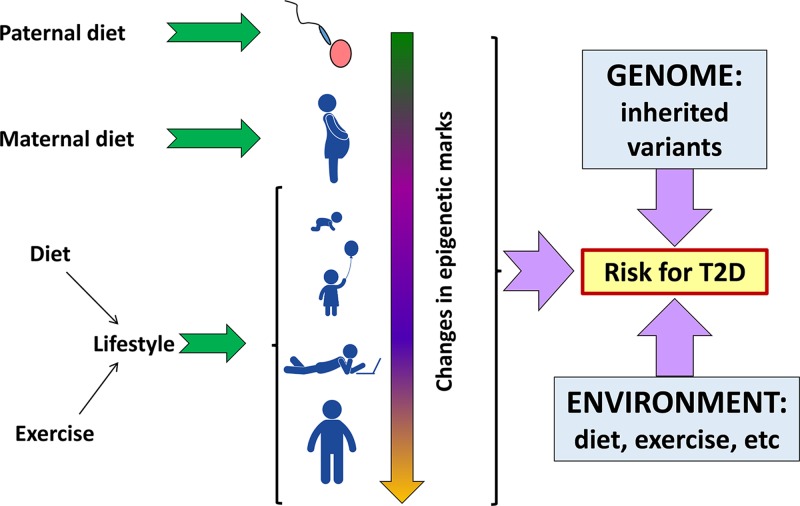
Epigenetics
-
- Lobo I. (2008) Genomic imprinting and patterns of disease inheritance. Nat. Education 1, 66
-
- Venturá-Junca P., Irarrázaval I., Rolle A.J., Gutiérrez J.I., Moreno R.D. and Santos M.J. (2015) In vitro fertilization (IVF) in mammals: epigenetic and developmental alterations. Scientific and bioethical implications for IVF in humans. Biol. Res. 48:, 68 10.1186/s40659-015-0059-y - DOI - PMC - PubMed
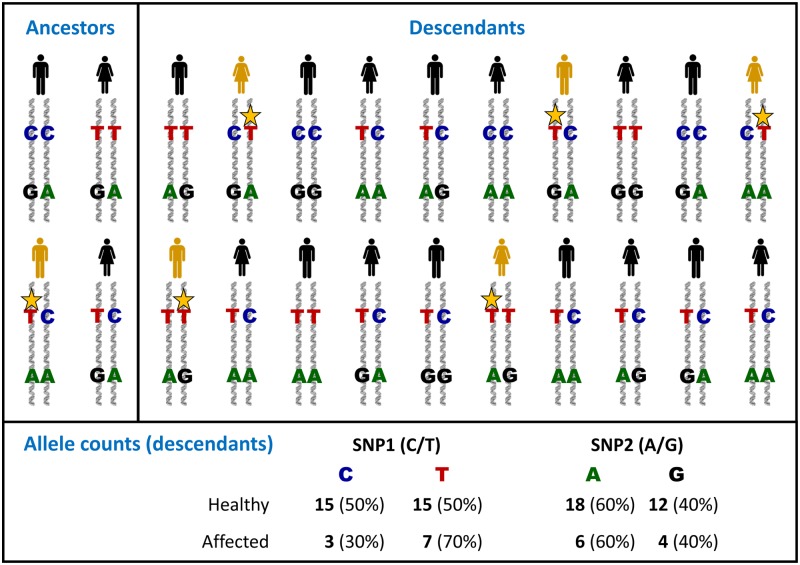
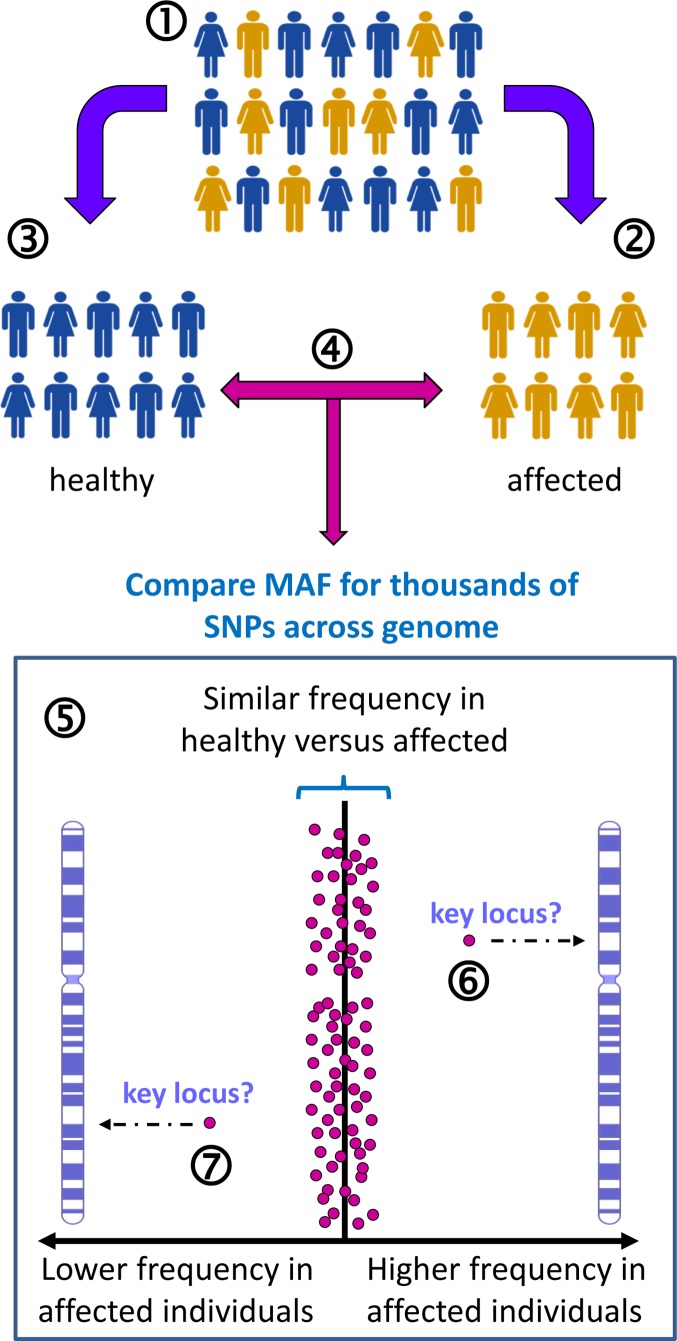
Complex disorders
-
- Chial H. and Craig J. (2008) Genome-wide association studies (GWAS) and obesity. Nat. Education 1, 80
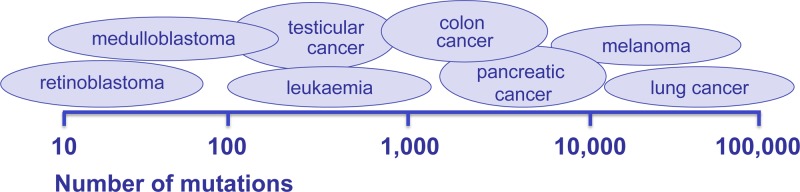
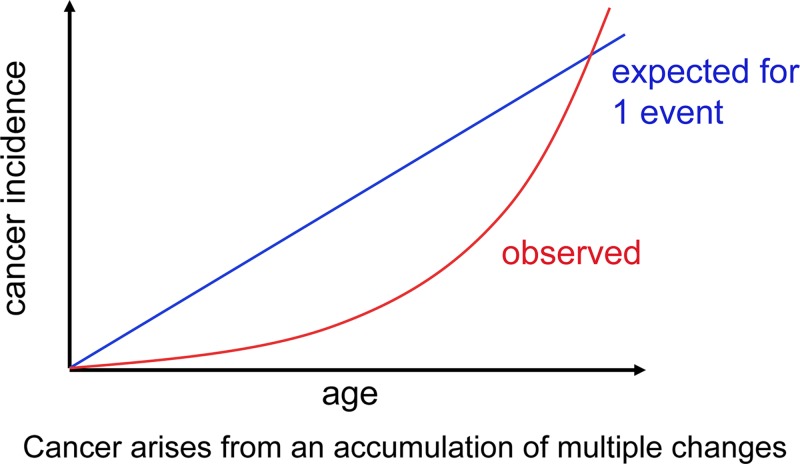
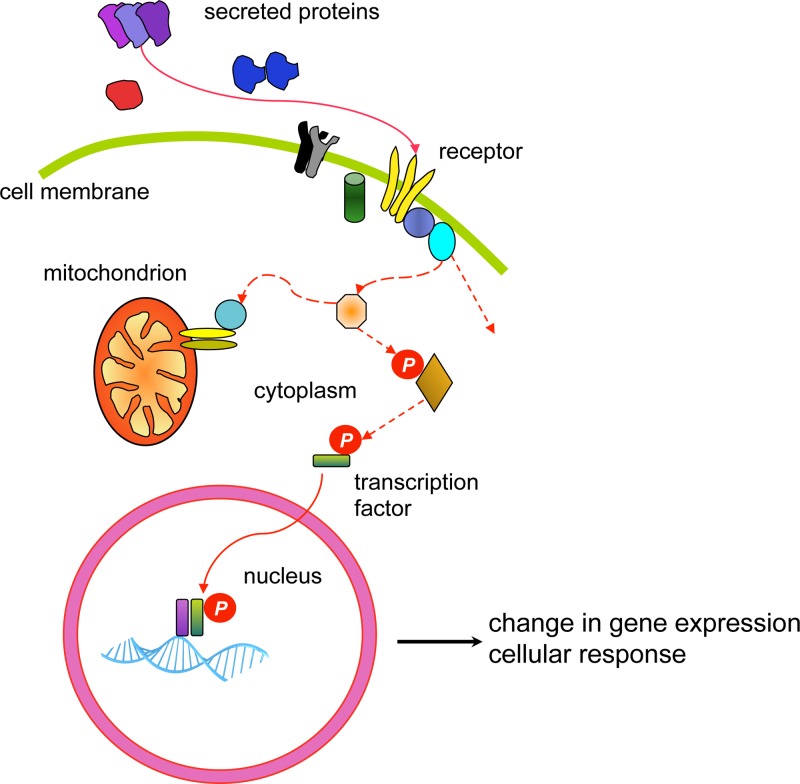
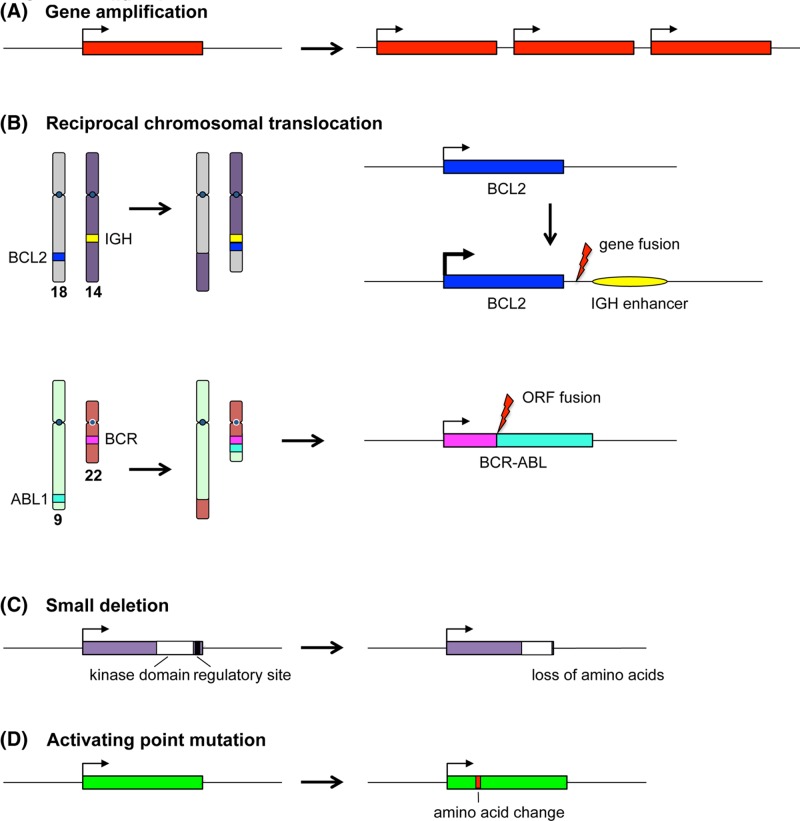
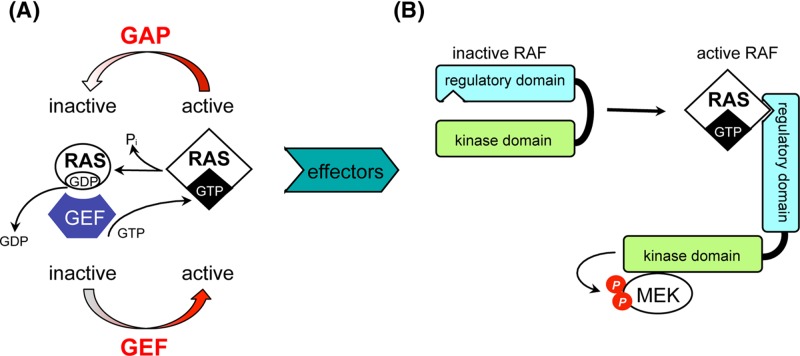
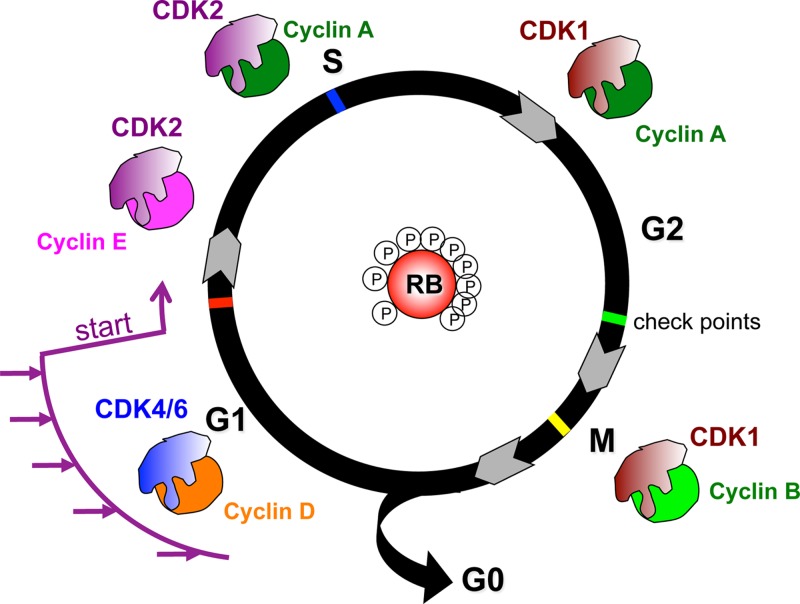
Cancer: mutation and epigenetics
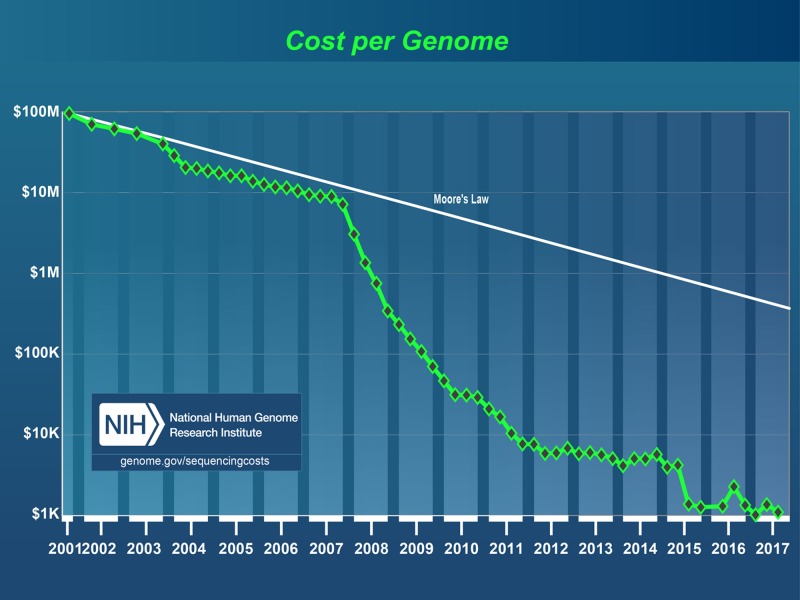
Genomics
-
- Genomics England, 100,000 genomes project, 2018. https://www.genomicsengland.co.uk/
-
- Scottish Genomes Partnership (2018), https://www.scottishgenomespartnership.org/
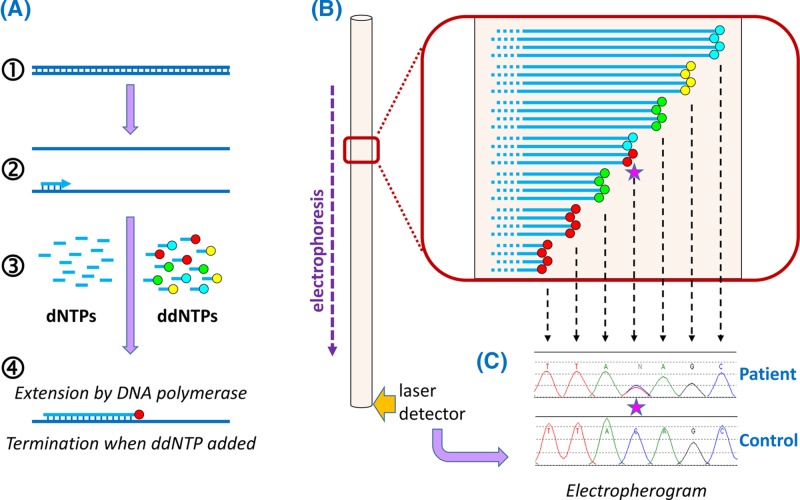
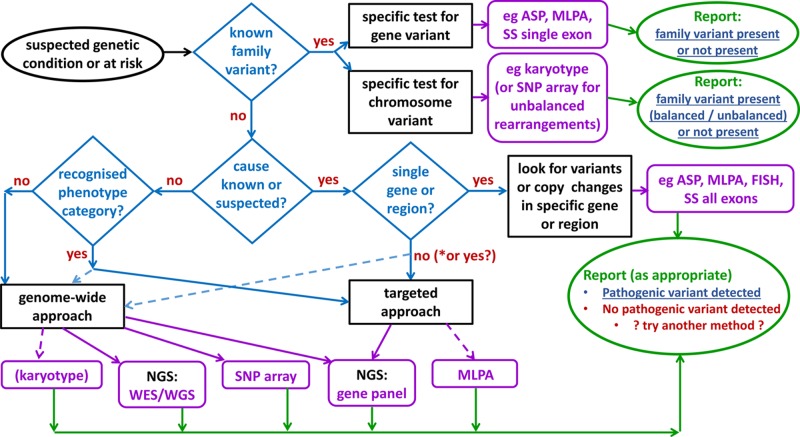
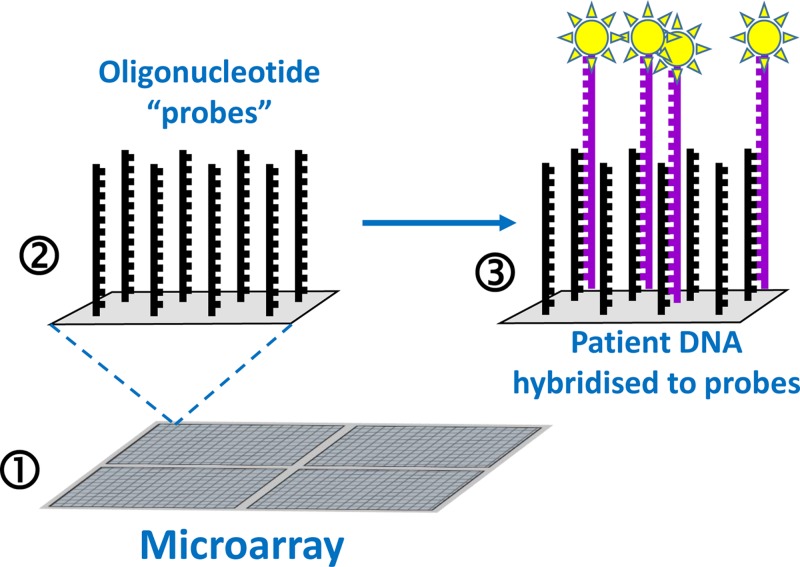
Genetic testing in the diagnostic laboratory
-
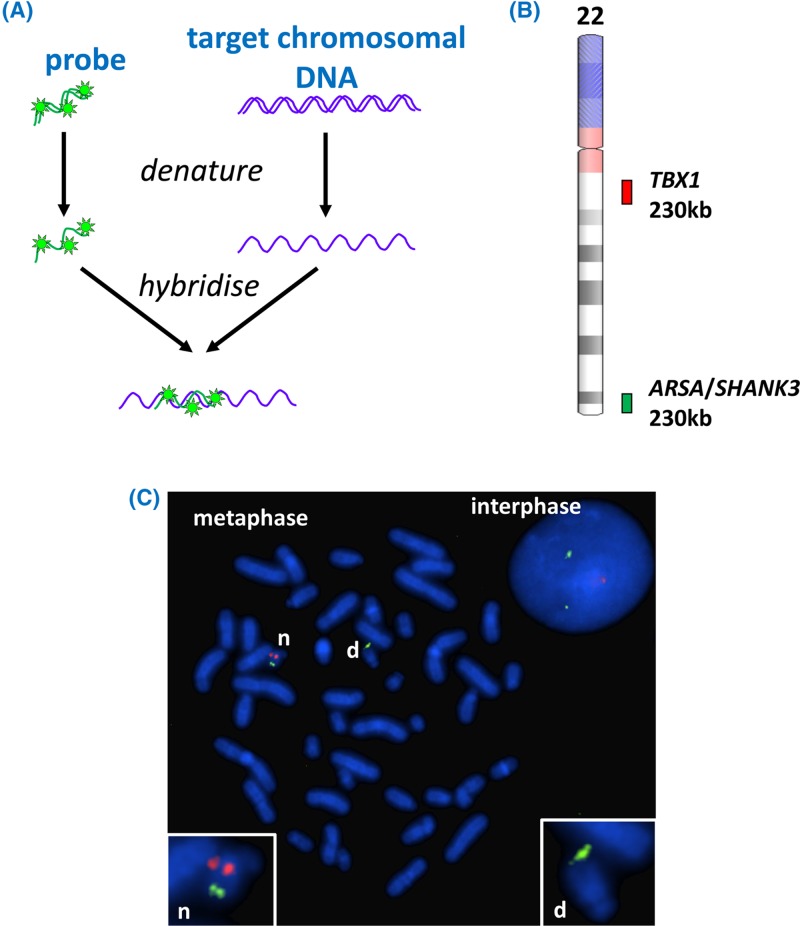
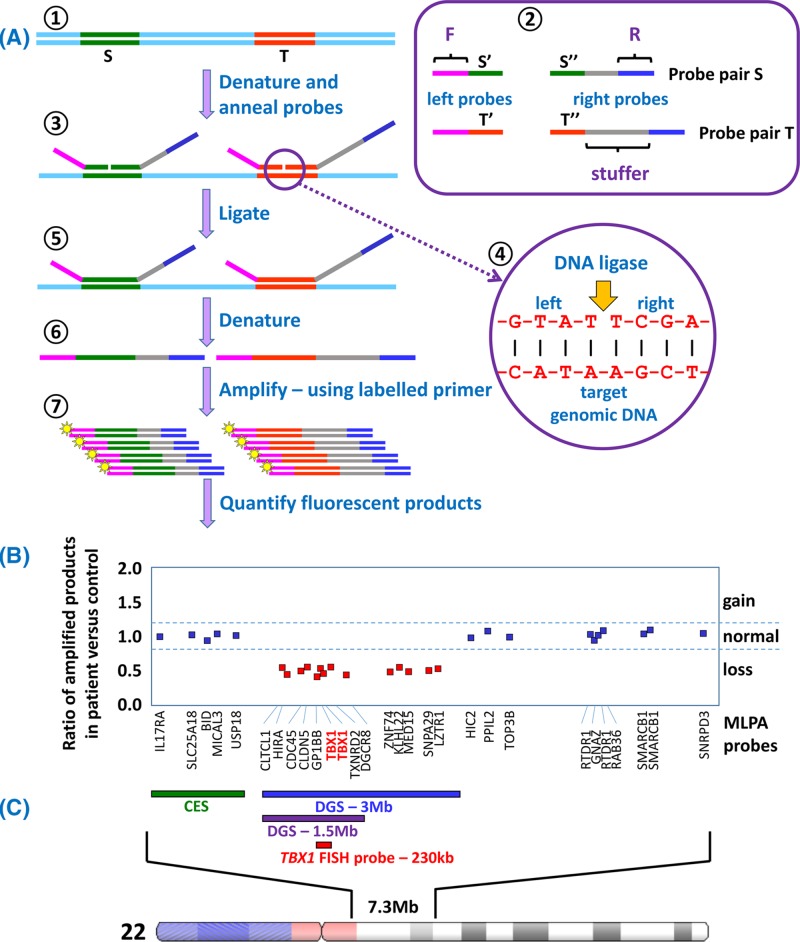
- Bishop R. (2010) Applications of fluorescence in situ hybridization (FISH) in detecting genetic aberrations of medical significance. Biosci. Horiz. 3, 85–95, 10.1093/biohorizons/hzq009 - DOI
-
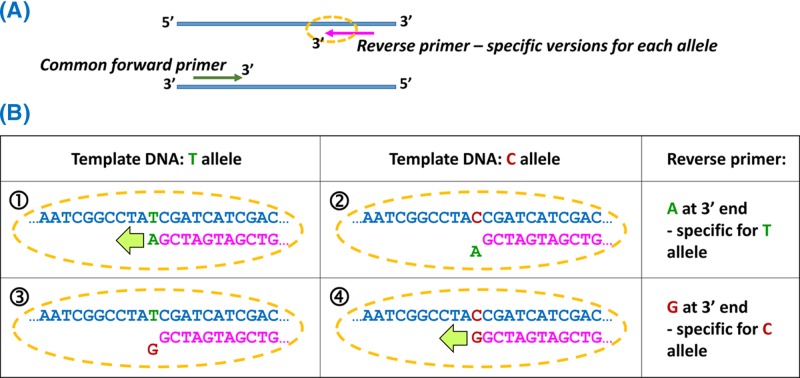
- Kchouk M., Gibrat J.-F. and Elloumi M. (2017) Generations of sequencing technologies: from first to next generation. Biol. Med. (Aligarh) 9, 395 10.4172/0974-8369.1000395 - DOI
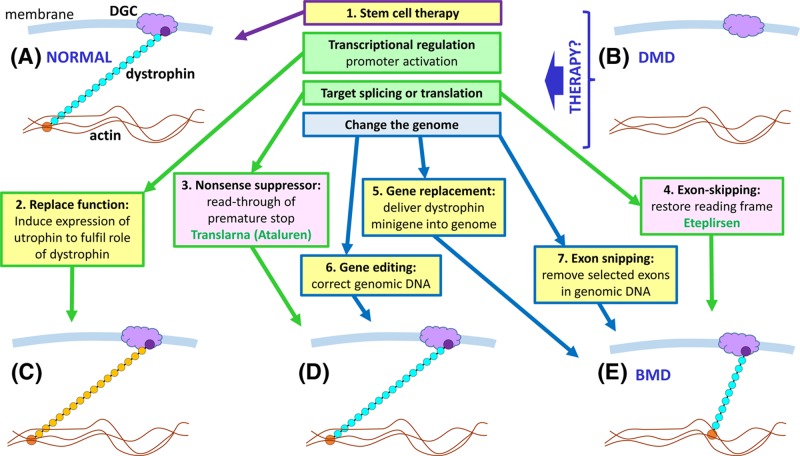
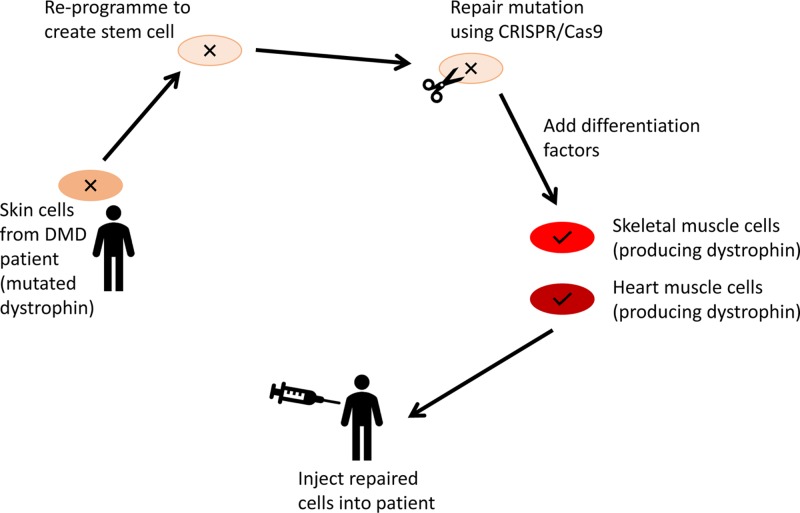
Diagnosis, management and therapy of genetic disease
-
- Lockyer E. (2016) The potential of CRISPR-Cas9 for treating genetic disorders. Biosci. Horiz. 9, 10.1093/biohorizons/hzw012 - DOI
Challenges in delivering a genetics service
-
- Blashki G., Metcalfe S. and Emery J. (2014) Genetics in general practice. Aust. Fam. Phys. 43, 428–431 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical