

Therapeutic strategies for sickle cell disease: towards a multi-agent approach
- PMID: 30514970
- PMCID: PMC6645400
- DOI: 10.1038/s41573-018-0003-2
Therapeutic strategies for sickle cell disease: towards a multi-agent approach
Abstract
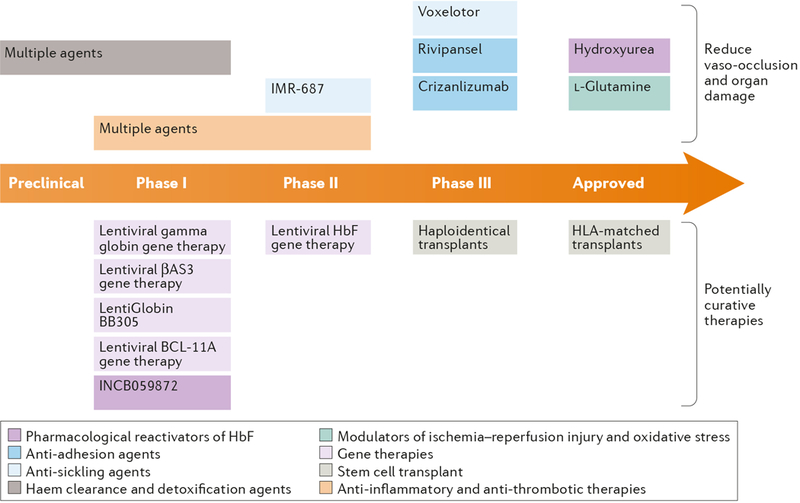
For over 100 years, clinicians and scientists have been unravelling the consequences of the A to T substitution in the β-globin gene that produces haemoglobin S, which leads to the systemic manifestations of sickle cell disease (SCD), including vaso-occlusion, anaemia, haemolysis, organ injury and pain. However, despite growing understanding of the mechanisms of haemoglobin S polymerization and its effects on red blood cells, only two therapies for SCD - hydroxyurea and L-glutamine - are approved by the US Food and Drug Administration. Moreover, these treatment options do not fully address the manifestations of SCD, which arise from a complex network of interdependent pathophysiological processes. In this article, we review efforts to develop new drugs targeting these processes, including agents that reactivate fetal haemoglobin, anti-sickling agents, anti-adhesion agents, modulators of ischaemia-reperfusion and oxidative stress, agents that counteract free haemoglobin and haem, anti-inflammatory agents, anti-thrombotic agents and anti-platelet agents. We also discuss gene therapy, which holds promise of a cure, although its widespread application is currently limited by technical challenges and the expense of treatment. We thus propose that developing systems-oriented multi-agent strategies on the basis of SCD pathophysiology is needed to improve the quality of life and survival of people with SCD.
Figures



References
-
- Platt OS et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death [see comments]. New England Journal of Medicine 330, 1639–1644 (1994). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
