

IVIg and LPS Co-stimulation Induces IL-10 Production by Human Monocytes, Which Is Compromised by an FcγRIIA Disease-Associated Gene Variant
- PMID: 30515163
- PMCID: PMC6255983
- DOI: 10.3389/fimmu.2018.02676
IVIg and LPS Co-stimulation Induces IL-10 Production by Human Monocytes, Which Is Compromised by an FcγRIIA Disease-Associated Gene Variant
Abstract
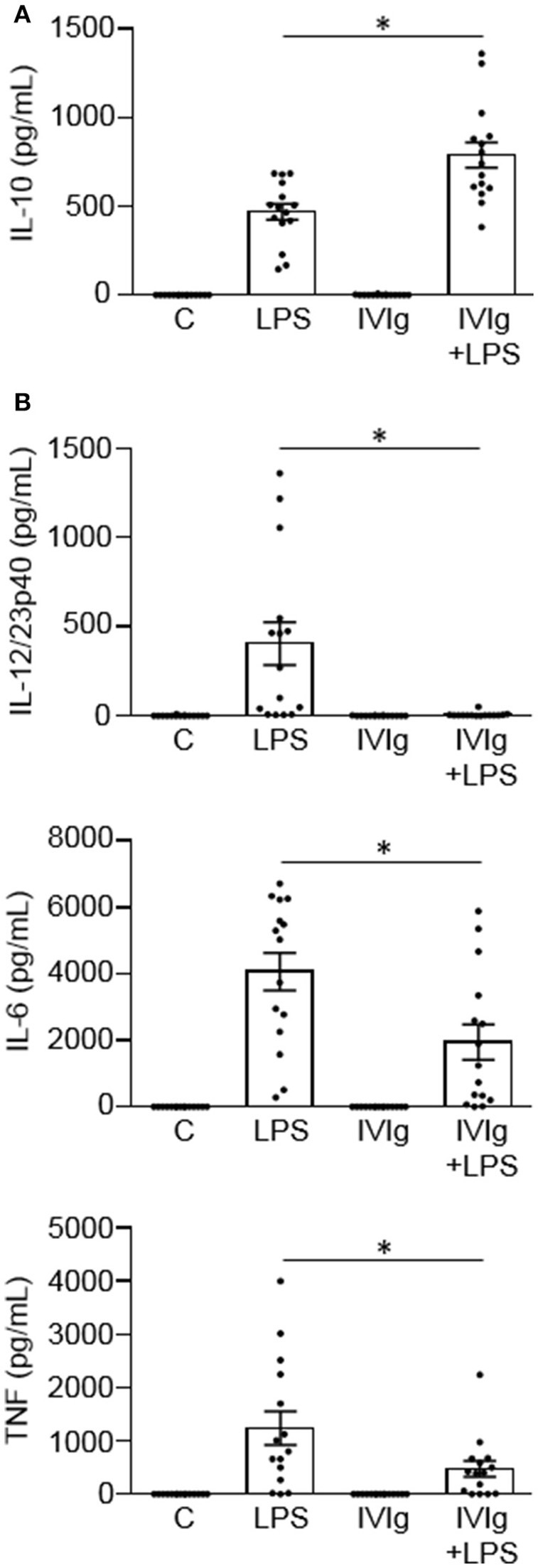
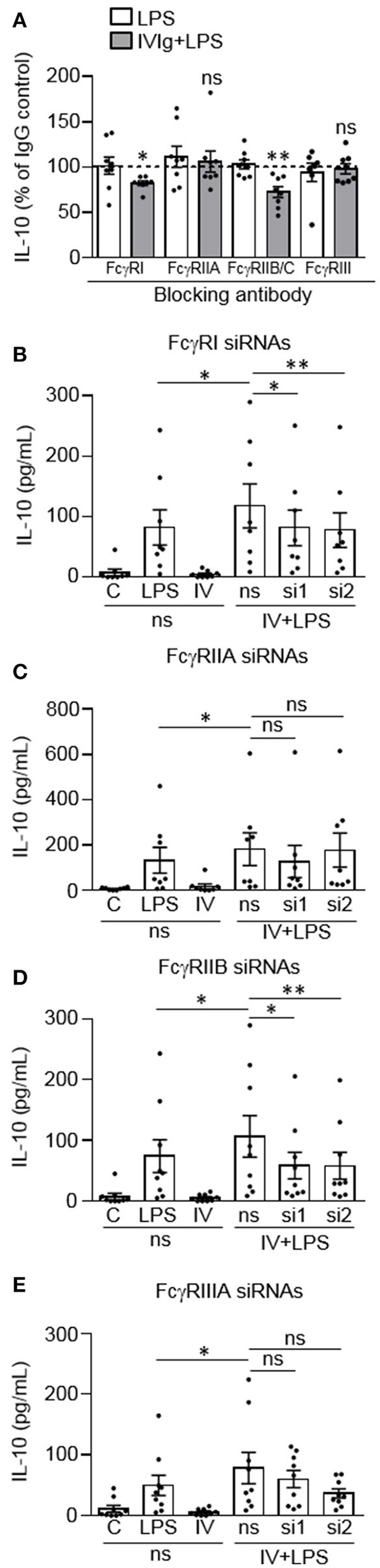
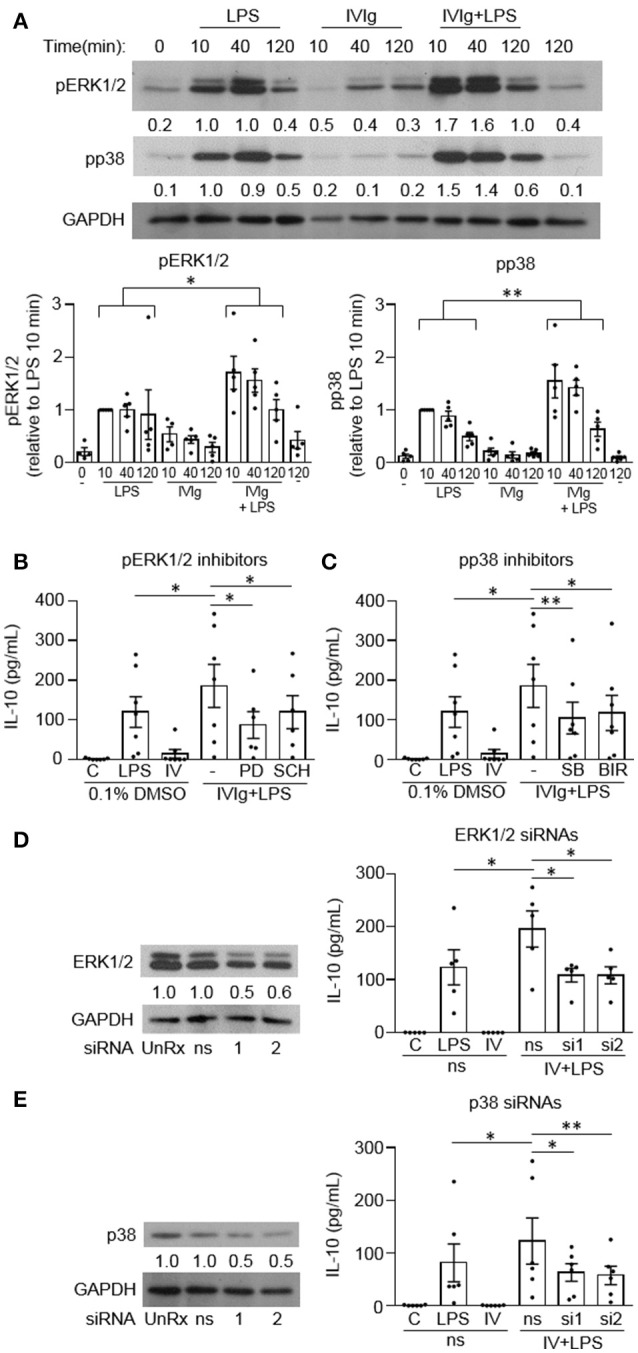
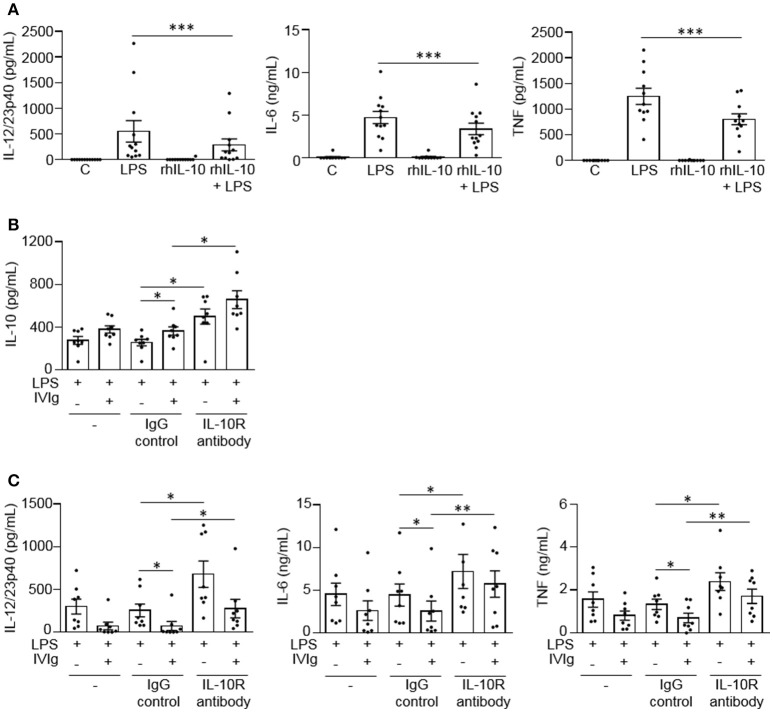
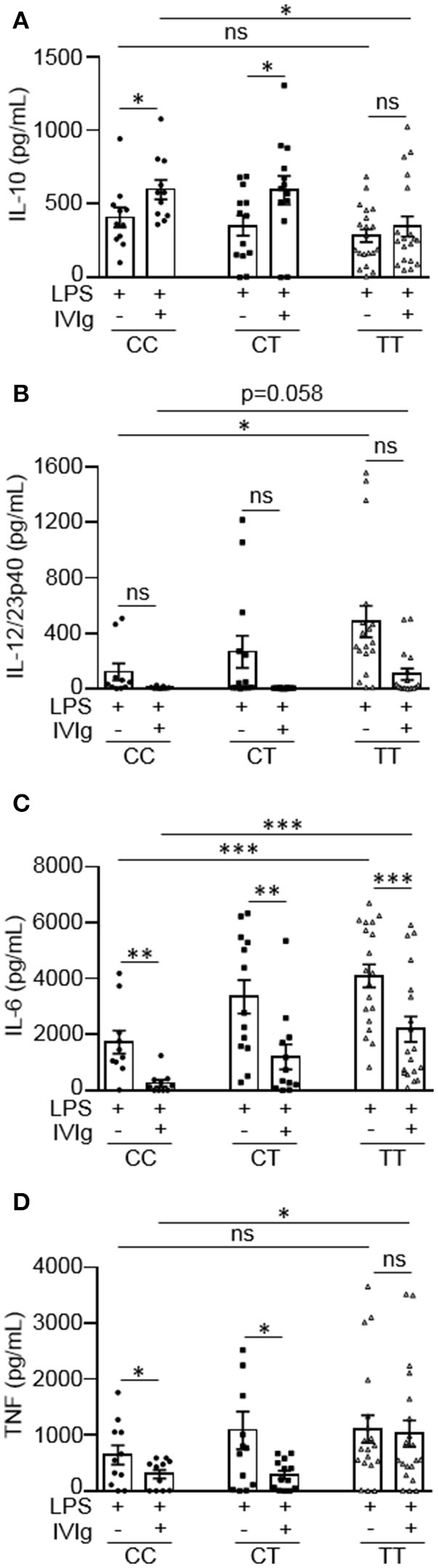
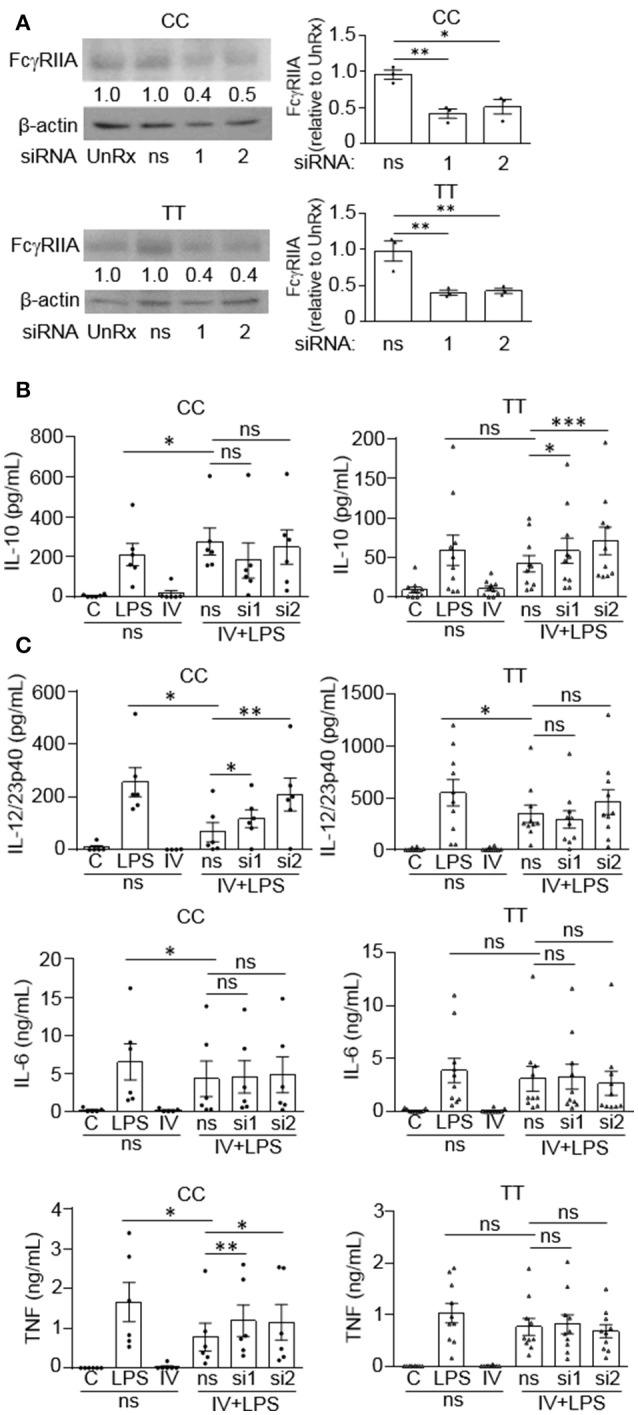
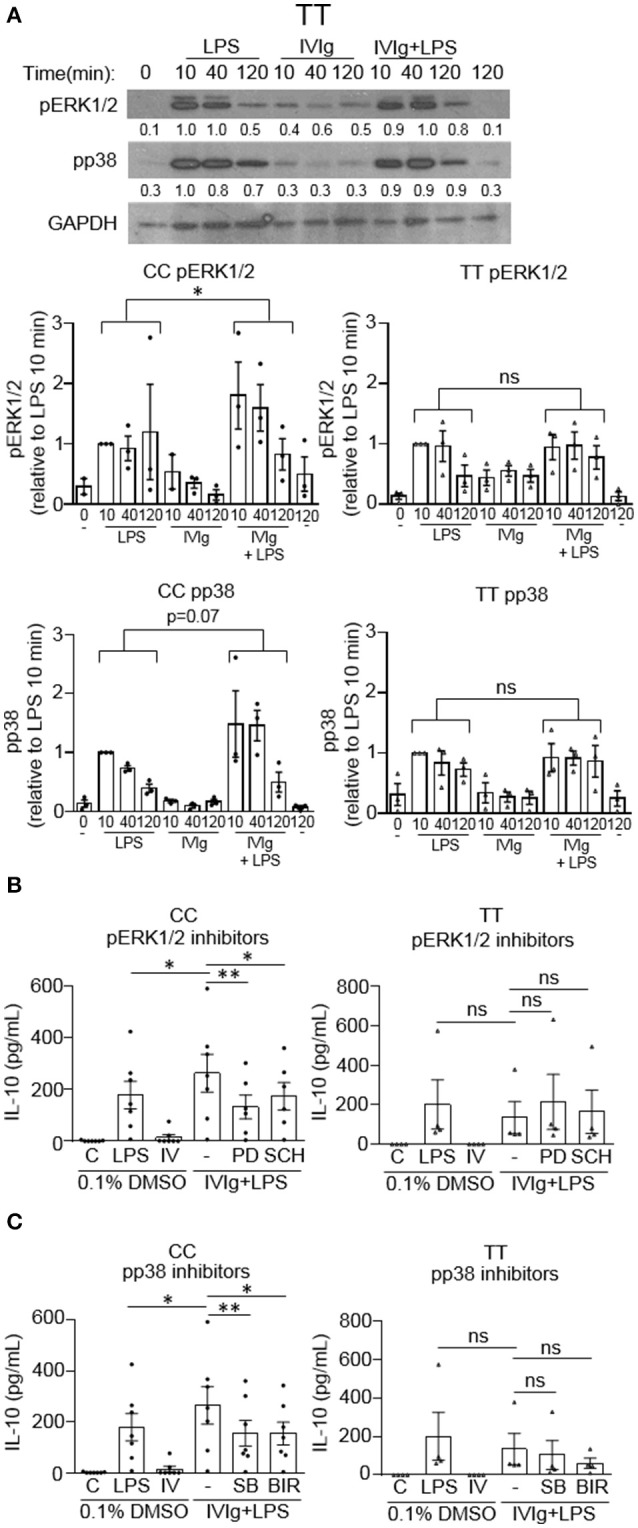
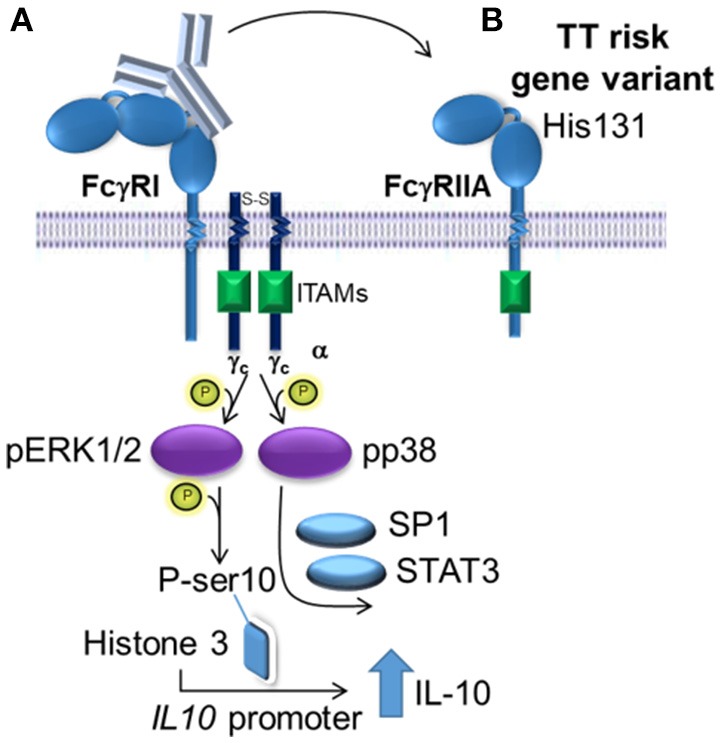
Intravenous Immunoglobulin (IVIg) is used to treat autoimmune or inflammatory diseases, but its mechanism of action is not completely understood. We asked whether IVIg can induce interleukin-10 (IL-10) and reduce pro-inflammatory cytokine production in human monocytes, and whether this response is reduced in monocytes from people with an Fcγ receptor IIA (FcγRIIA) gene variant, which is associated with increased risk of inflammatory diseases and poor response to antibody-based biological therapy. IVIg increased IL-10 production and reduced pro-inflammatory cytokine production in response to bacterial lipopolysaccharide (LPS), which required FcγRI and FcγRIIB and activation of MAPKs, extracellular signal-regulated kinase 1/2 (ERK1/2), and p38. IL-10 production was lower and pro-inflammatory cytokine production was higher in monocytes from people with the FcγRIIA risk variant and the risk variant prevented IL-10 production in response to (IVIg+LPS). Finally, we show that IVIg did not induce MAPK activation in monocytes from people with the risk variant. Our results demonstrate that IVIg can skew human monocytes to an anti-inflammatory, IL-10-producing activation state, which is compromised in monocytes from people with the FcγRIIA risk variant. This research has profound implications for the use of IVIg because 25% of the population is homozygous for the FcγRIIA risk variant and its efficacy may be reduced in those individuals. In addition, this research may be useful to develop new therapeutic strategies to replace IVIg by cross-linking FcγRIs and FcγRIIBs to promote anti-inflammatory macrophage activation, independent of the FcγRIIA genotype.
Keywords: ERK; FcγRIIA; IL-10; IVIg; MAPK; monocyte; p38; rs1801274.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous