

Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator
- PMID: 30517868
- PMCID: PMC6342284
- DOI: 10.1016/j.celrep.2018.11.035
Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator
Abstract
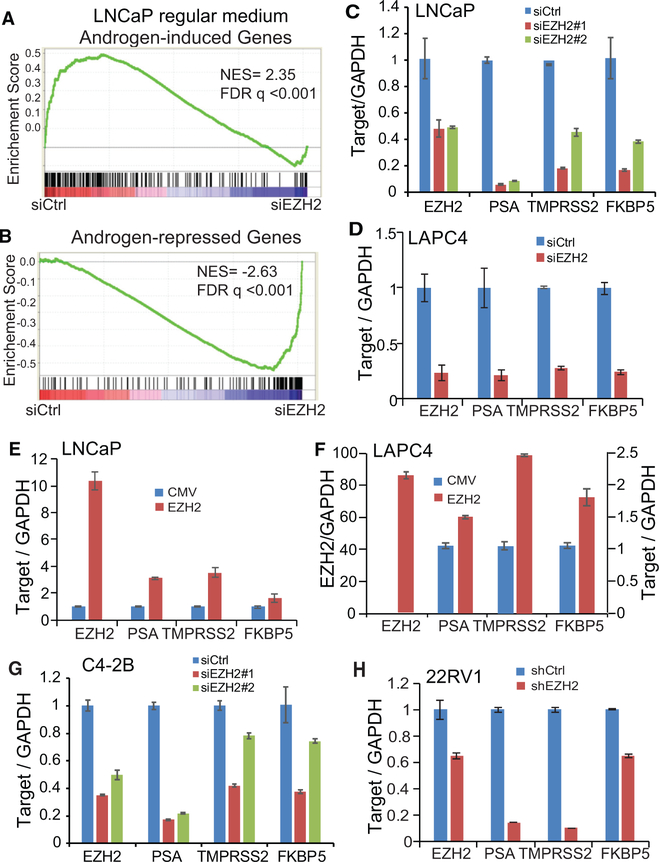
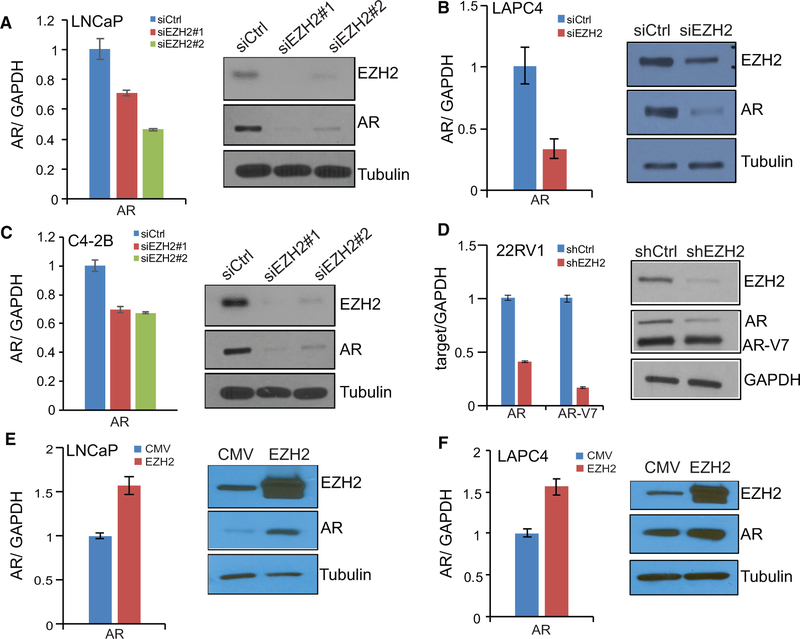
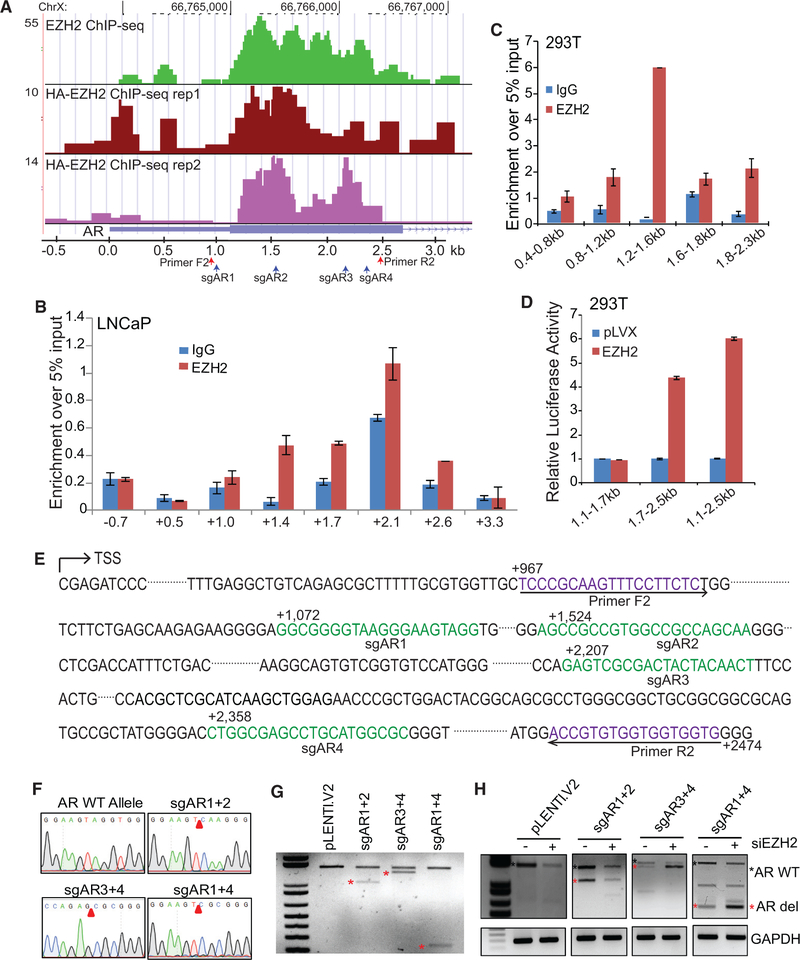
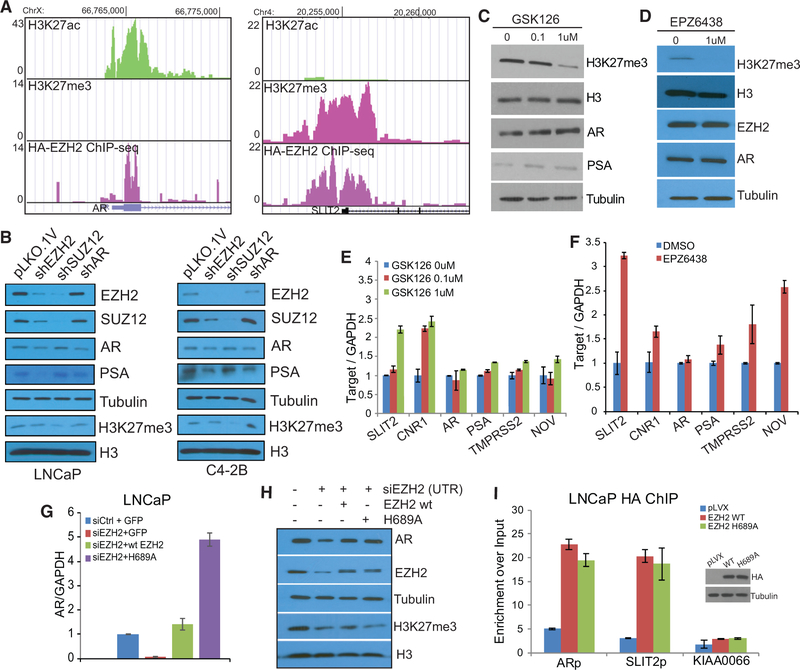
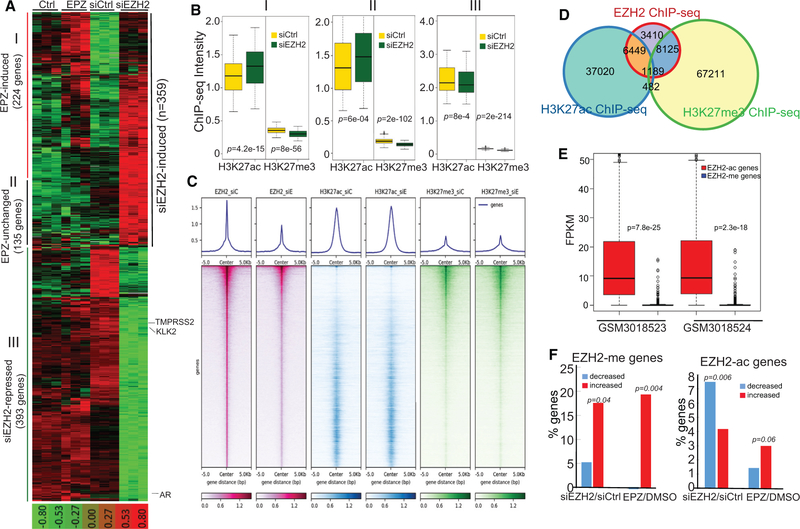
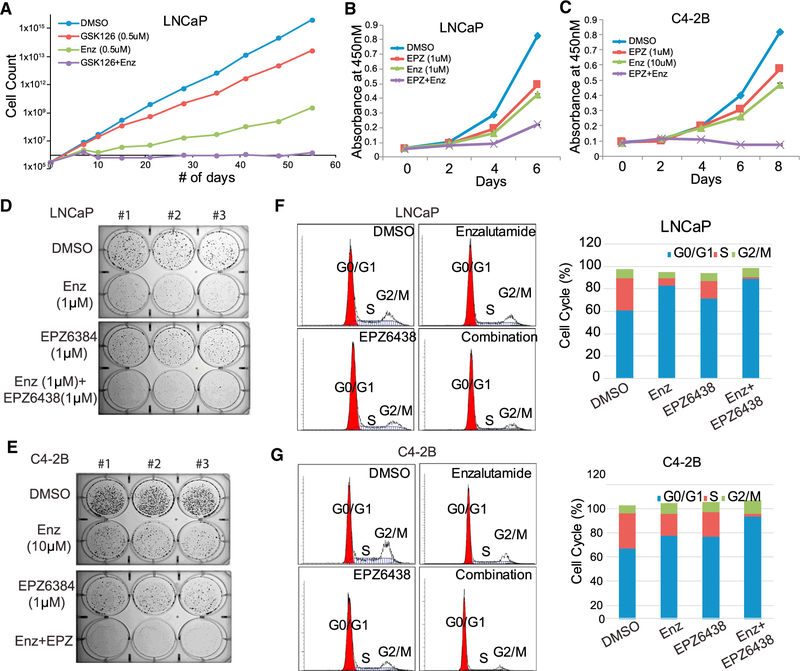
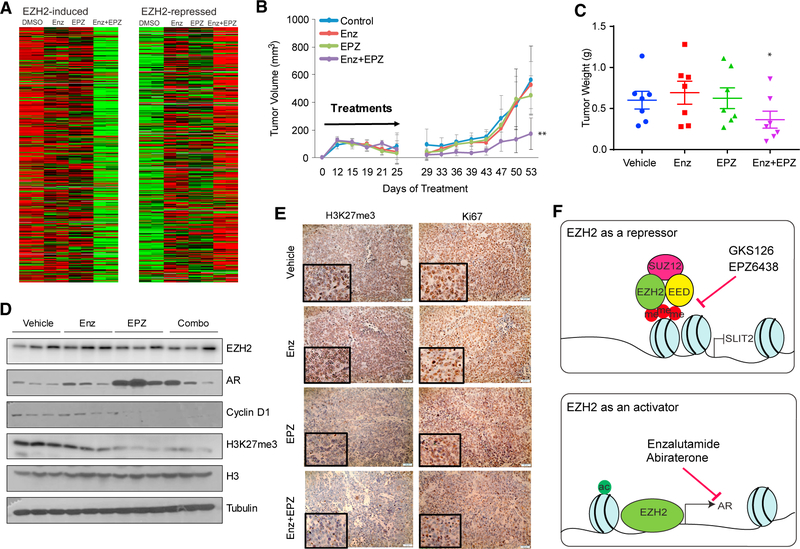
Enhancer of Zeste 2 (EZH2) is the enzymatic subunit of Polycomb Repressive Complex 2 (PRC2), which catalyzes histone H3 lysine 27 trimethylation (H3K27me3) at target promoters for gene silencing. Here, we report that EZH2 activates androgen receptor (AR) gene transcription through direct occupancy at its promoter. Importantly, this activating role of EZH2 is independent of PRC2 and its methyltransferase activities. Genome-wide assays revealed extensive EZH2 occupancy at promoters marked by either H3K27ac or H3K27me3, leading to gene activation or repression, respectively. Last, we demonstrate enhanced efficacy of enzymatic EZH2 inhibitors when used in combination with AR antagonists in blocking the dual roles of EZH2 and suppressing prostate cancer progression in vitro and in vivo. Taken together, our study reports EZH2 as a transcriptional activator, a key target of which is AR, and suggests a drug-combinatory approach to treat advanced prostate cancer.
Keywords: AR antagonist enzalutamide; ChIP-seq; EPZ-6438; GSK126; androgen receptor inhibitor; enzymatic EZH2 inhibitor; epigenetic silencing; transcription activator.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
References
-
- Höller M, Westin G, Jiricny J, and Schaffner W (1988). Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes Dev. 2, 1127–1135. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
