

Converter domain mutations in myosin alter structural kinetics and motor function
- PMID: 30518549
- PMCID: PMC6364761
- DOI: 10.1074/jbc.RA118.006128
Converter domain mutations in myosin alter structural kinetics and motor function
Abstract
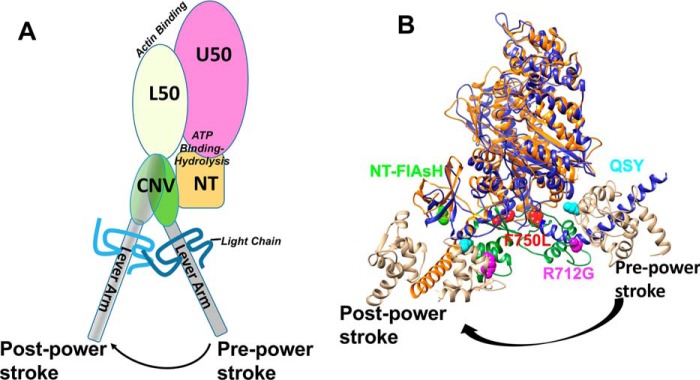
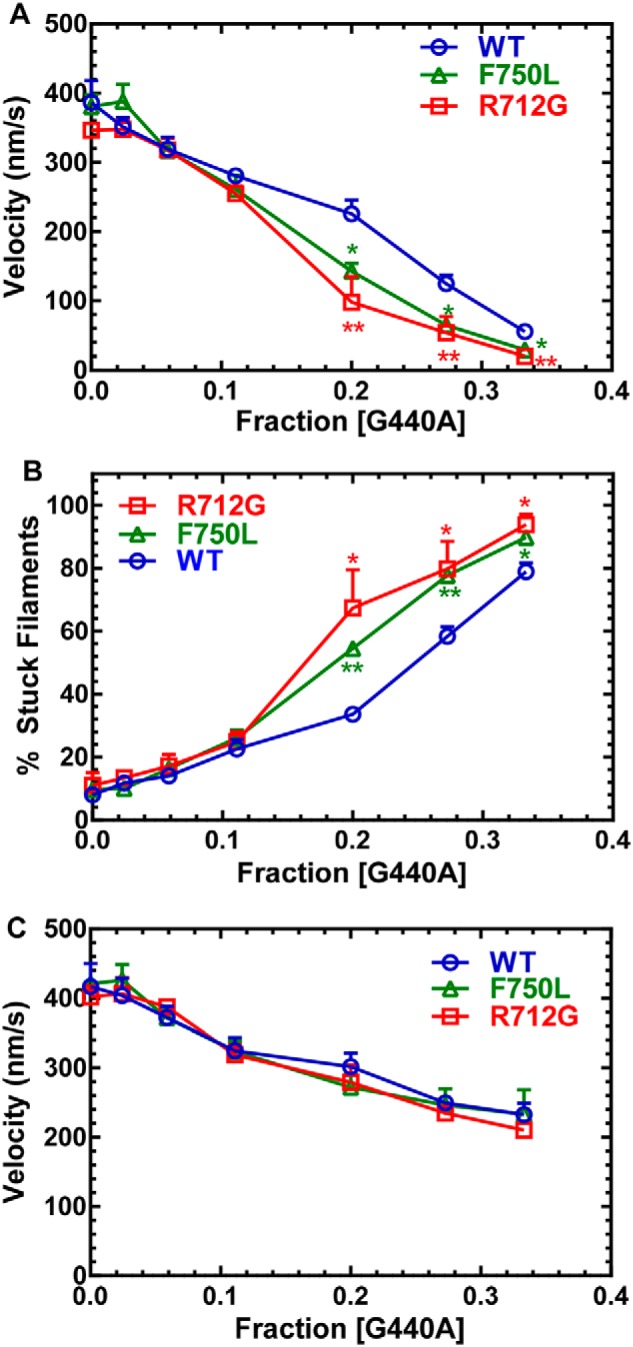
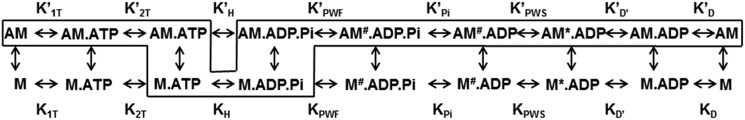
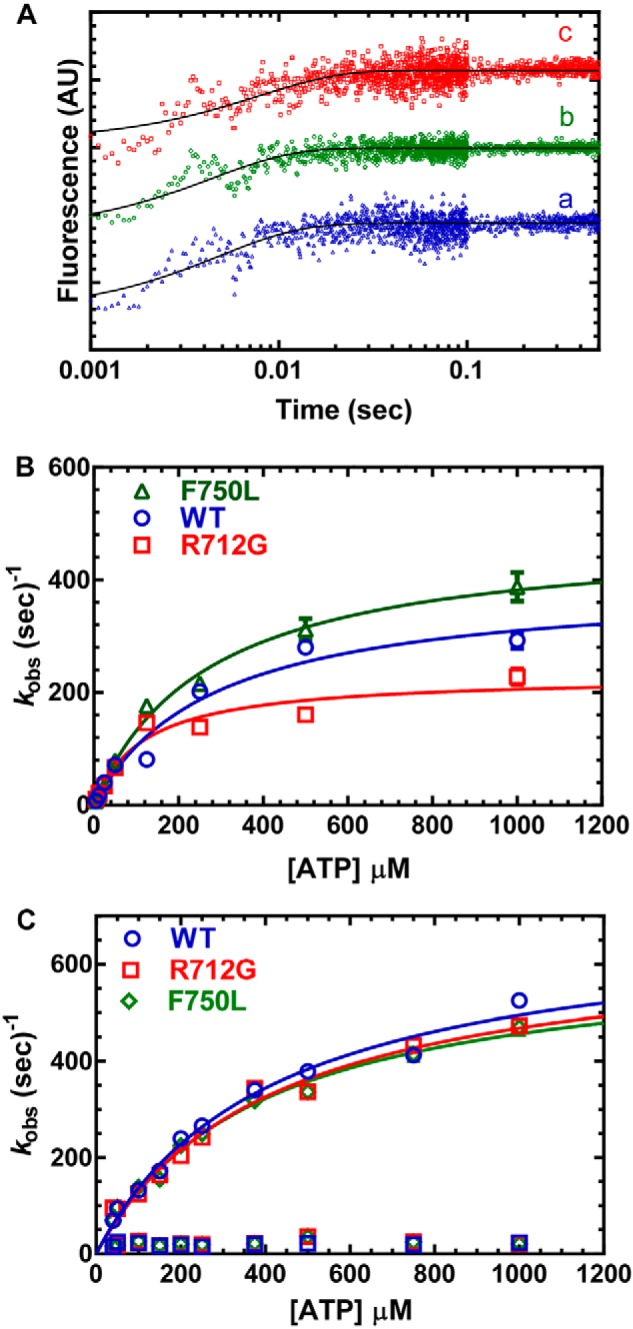
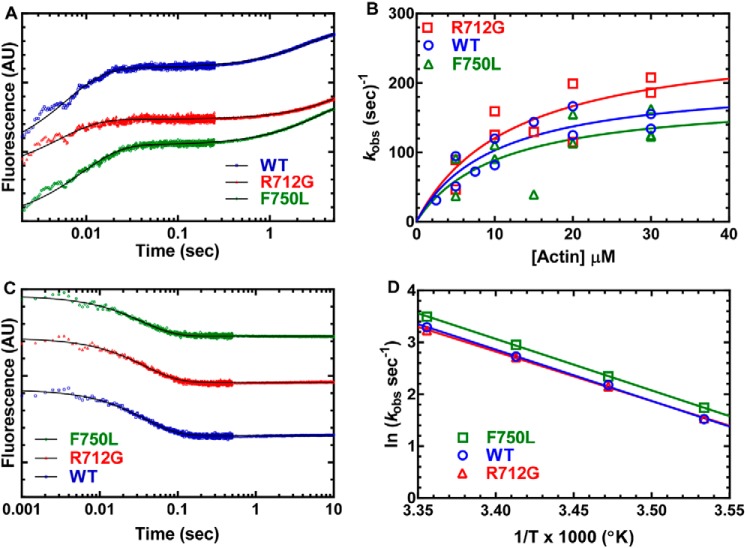
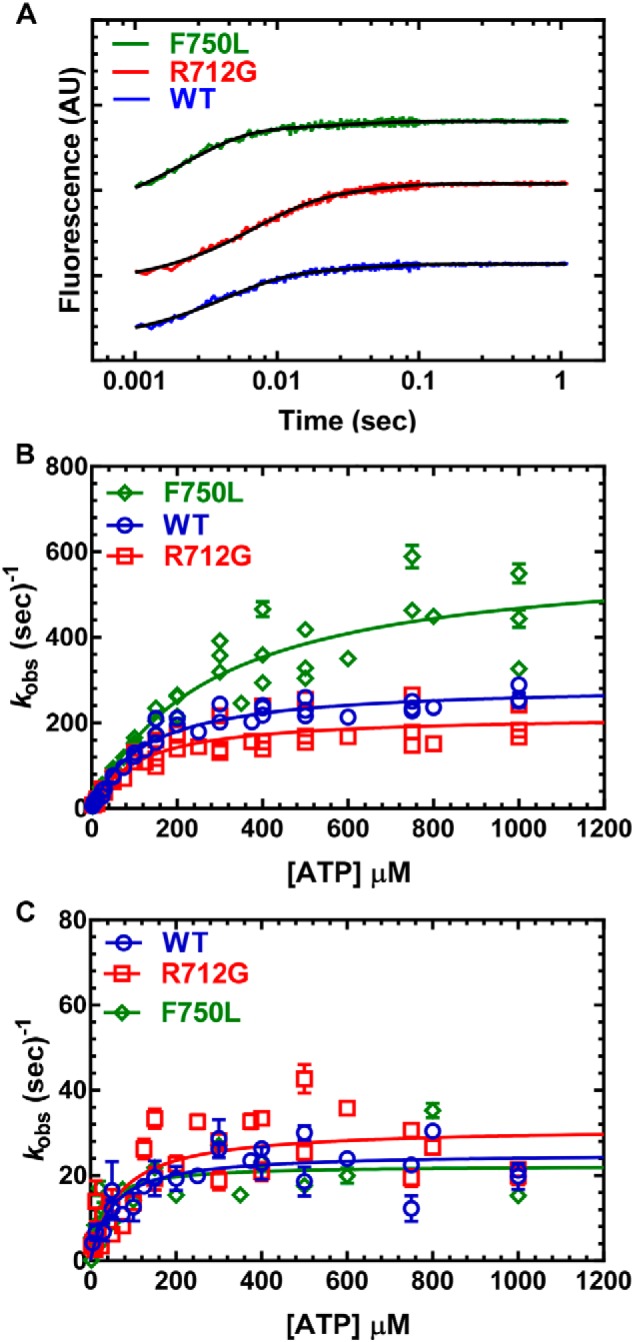
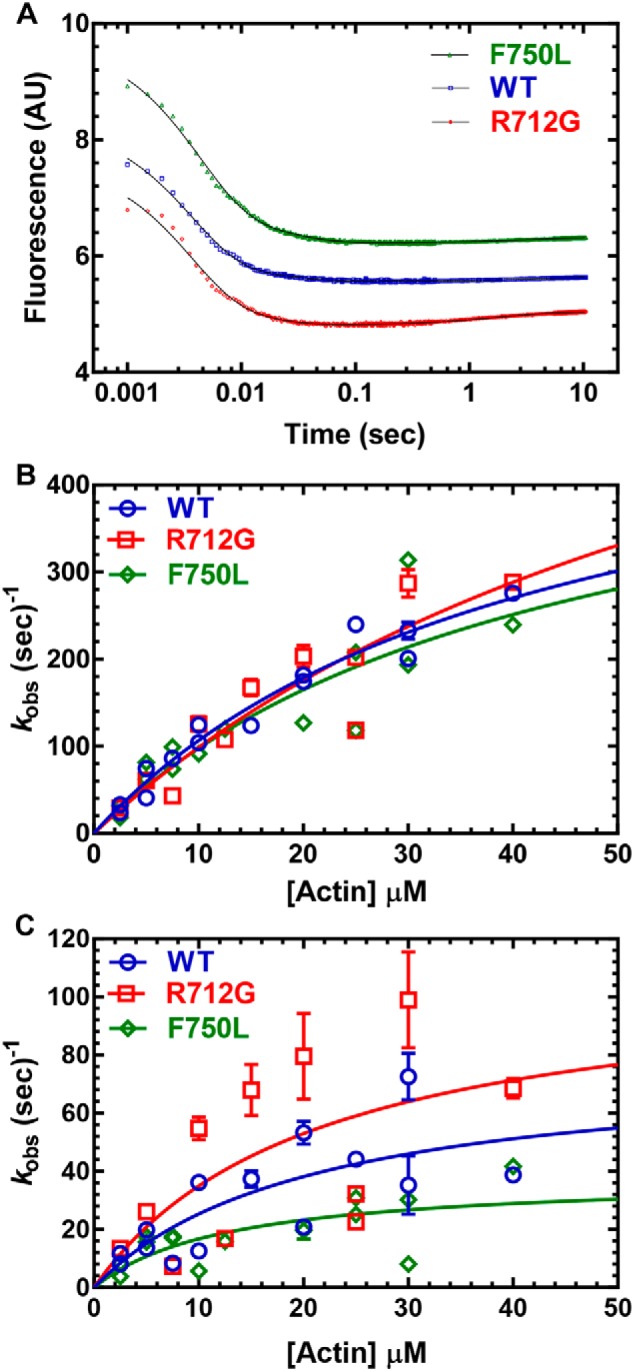
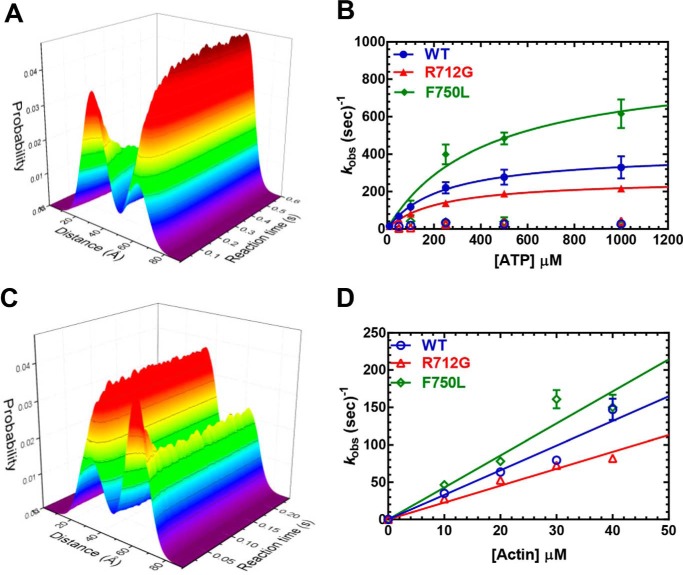
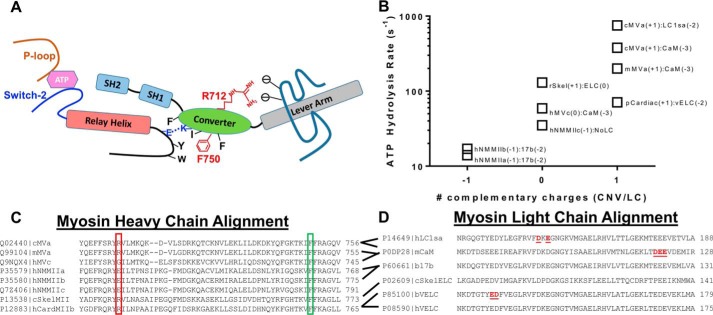
Myosins are molecular motors that use a conserved ATPase cycle to generate force. We investigated two mutations in the converter domain of myosin V (R712G and F750L) to examine how altering specific structural transitions in the motor ATPase cycle can impair myosin mechanochemistry. The corresponding mutations in the human β-cardiac myosin gene are associated with hypertrophic and dilated cardiomyopathy, respectively. Despite similar steady-state actin-activated ATPase and unloaded in vitro motility-sliding velocities, both R712G and F750L were less able to overcome frictional loads measured in the loaded motility assay. Transient kinetic analysis and stopped-flow FRET demonstrated that the R712G mutation slowed the maximum ATP hydrolysis and recovery-stroke rate constants, whereas the F750L mutation enhanced these steps. In both mutants, the fast and slow power-stroke as well as actin-activated phosphate release rate constants were not significantly different from WT. Time-resolved FRET experiments revealed that R712G and F750L populate the pre- and post-power-stroke states with similar FRET distance and distance distribution profiles. The R712G mutant increased the mole fraction in the post-power-stroke conformation in the strong actin-binding states, whereas the F750L decreased this population in the actomyosin ADP state. We conclude that mutations in key allosteric pathways can shift the equilibrium and/or alter the activation energy associated with key structural transitions without altering the overall conformation of the pre- and post-power-stroke states. Thus, therapies designed to alter the transition between structural states may be able to rescue the impaired motor function induced by disease mutations.
Keywords: ATPase; actin; cardiomyopathy; fluorescence resonance energy transfer (FRET); mechanochemistry; motor protein; muscle; myosin; structural kinetics.
© 2019 Gunther et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Homburger J. R., Green E. M., Caleshu C., Sunitha M. S., Taylor R. E., Ruppel K. M., Metpally R. P., Colan S. D., Michels M., Day S. M., Olivotto I., Bustamante C. D., Dewey F. E., Ho C. Y., Spudich J. A., and Ashley E. A. (2016) Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. U.S.A. 113, 6701–6706 10.1073/pnas.1606950113 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
