

Prisoners of war - host adaptation and its constraints on virus evolution
- PMID: 30518814
- PMCID: PMC7097816
- DOI: 10.1038/s41579-018-0120-2
Prisoners of war - host adaptation and its constraints on virus evolution
Abstract
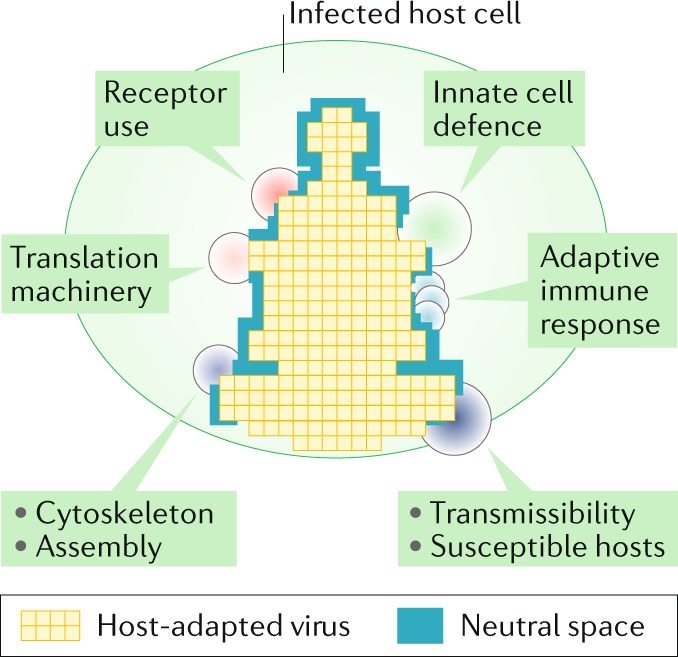
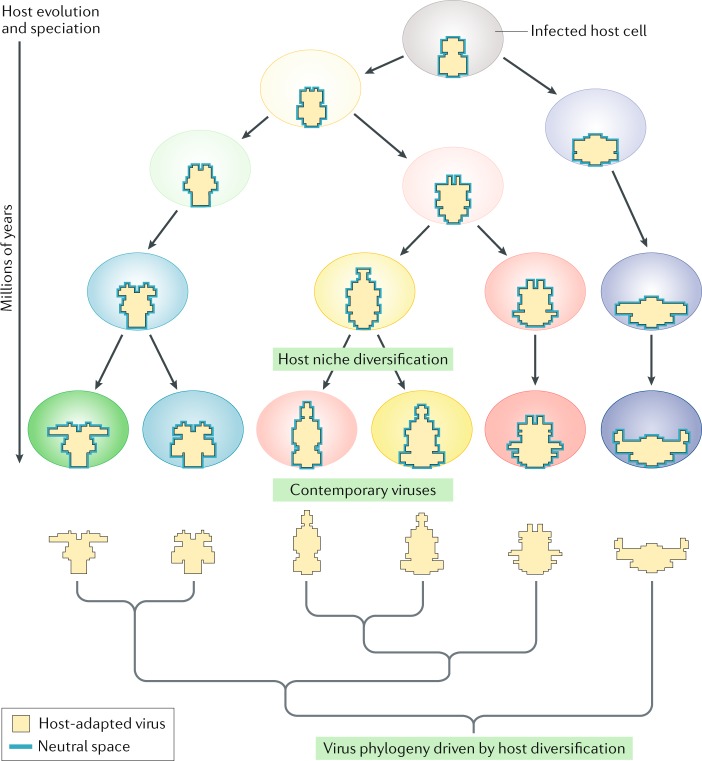
Recent discoveries of contemporary genotypes of hepatitis B virus and parvovirus B19 in ancient human remains demonstrate that little genetic change has occurred in these viruses over 4,500-6,000 years. Endogenous viral elements in host genomes provide separate evidence that viruses similar to many major contemporary groups circulated 100 million years ago or earlier. In this Opinion article, we argue that the extraordinary conservation of virus genome sequences is best explained by a niche-filling model in which fitness optimization is rapidly achieved in their specific hosts. Whereas short-term substitution rates reflect the accumulation of tolerated sequence changes within adapted genomes, longer-term rates increasingly resemble those of their hosts as the evolving niche moulds and effectively imprisons the virus in co-adapted virus-host relationships. Contrastingly, viruses that jump hosts undergo strong and stringent adaptive selection as they maximize their fit to their new niche. This adaptive capability may paradoxically create evolutionary stasis in long-term host relationships. While viruses can evolve and adapt rapidly, their hosts may ultimately shape their longer-term evolution.
Conflict of interest statement
The authors declare no competing interests.
Figures


Comment in
-
Reply to 'Evolutionary stasis of viruses?'.Nat Rev Microbiol. 2019 May;17(5):329-330. doi: 10.1038/s41579-019-0169-6. Nat Rev Microbiol. 2019. PMID: 30814681 No abstract available.
-
Evolutionary stasis of viruses?Nat Rev Microbiol. 2019 May;17(5):329. doi: 10.1038/s41579-019-0168-7. Nat Rev Microbiol. 2019. PMID: 30814682 No abstract available.
References
-
- Lemey P, Rambaut A, Pybus OG. HIV evolutionary dynamics within and among hosts. AIDS Rev. 2006;8:125–140. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
