

Single-cell mapping of lineage and identity in direct reprogramming
- PMID: 30518857
- PMCID: PMC6635140
- DOI: 10.1038/s41586-018-0744-4
Single-cell mapping of lineage and identity in direct reprogramming
Abstract
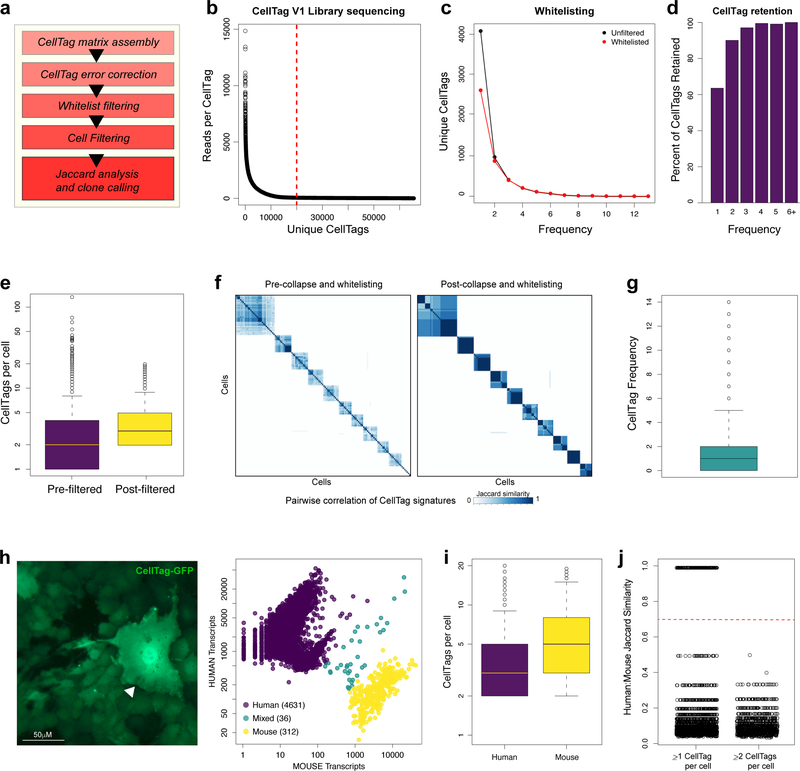
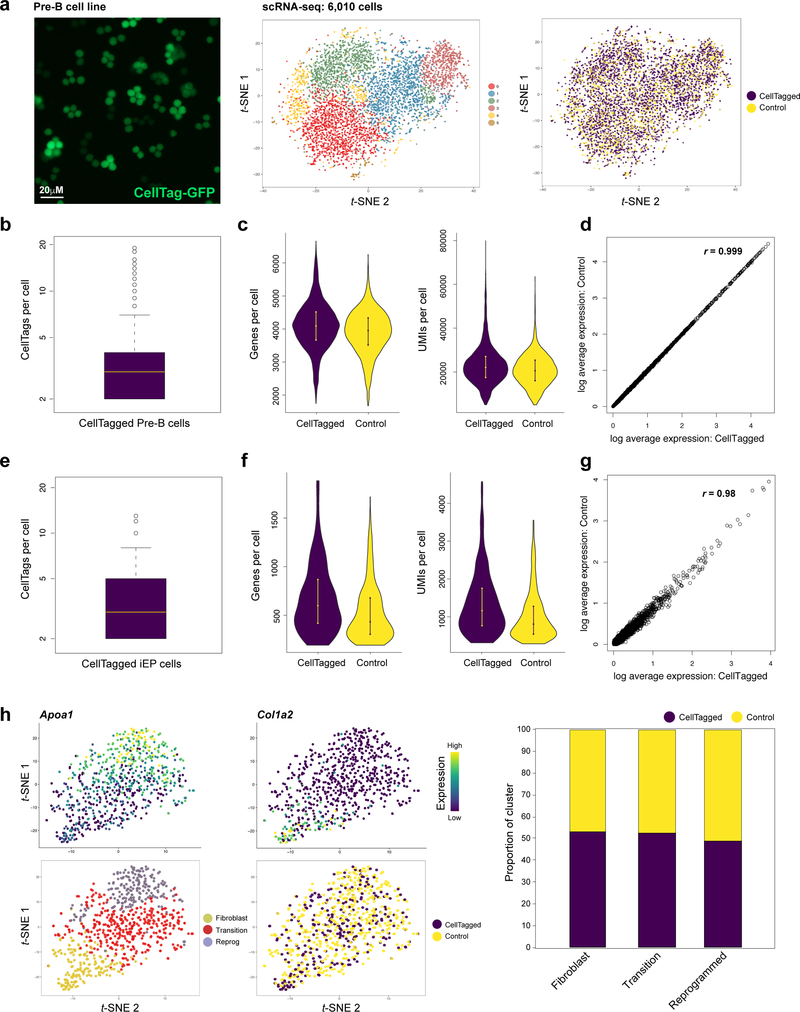
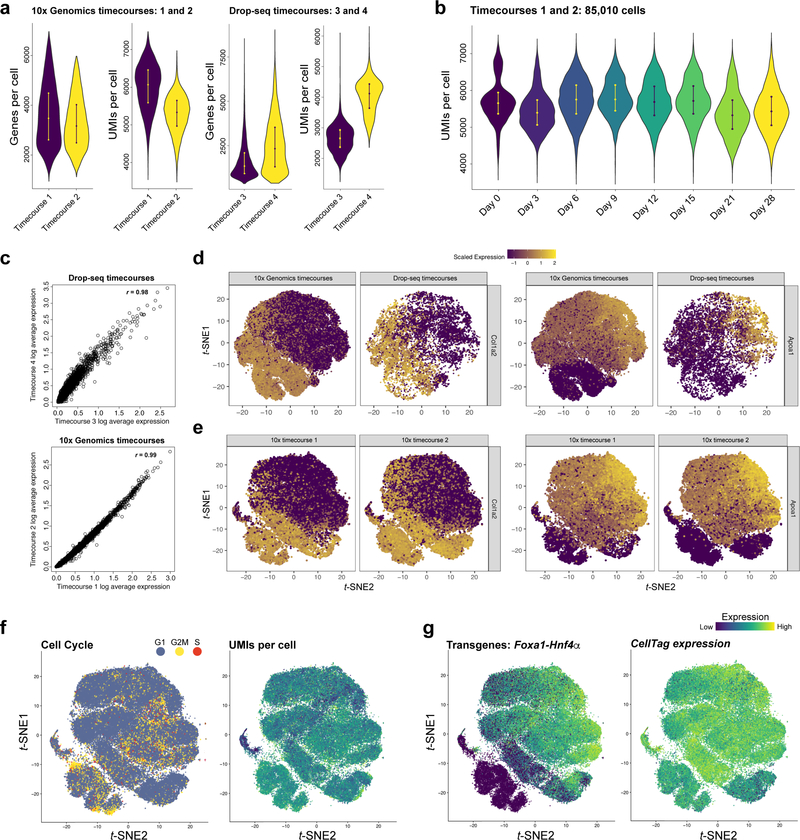
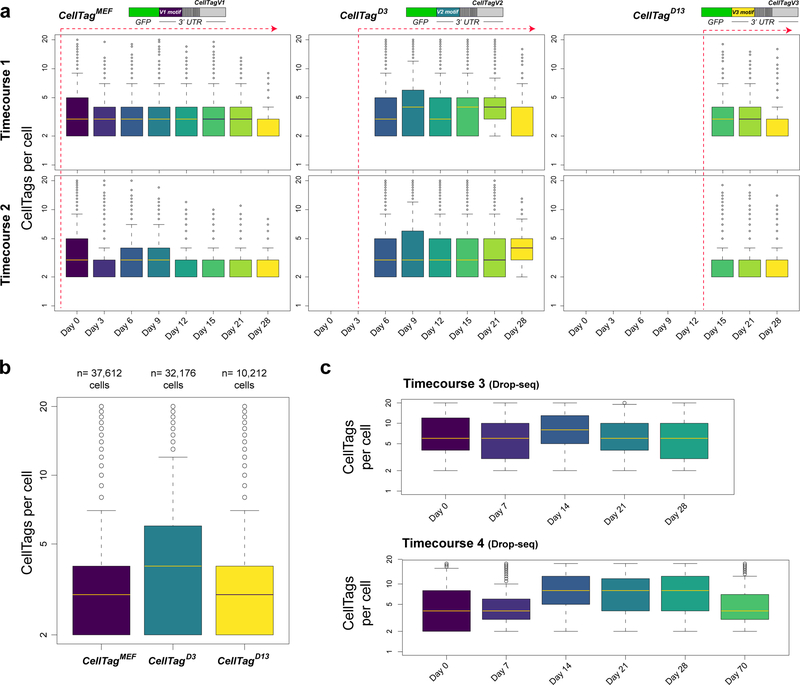
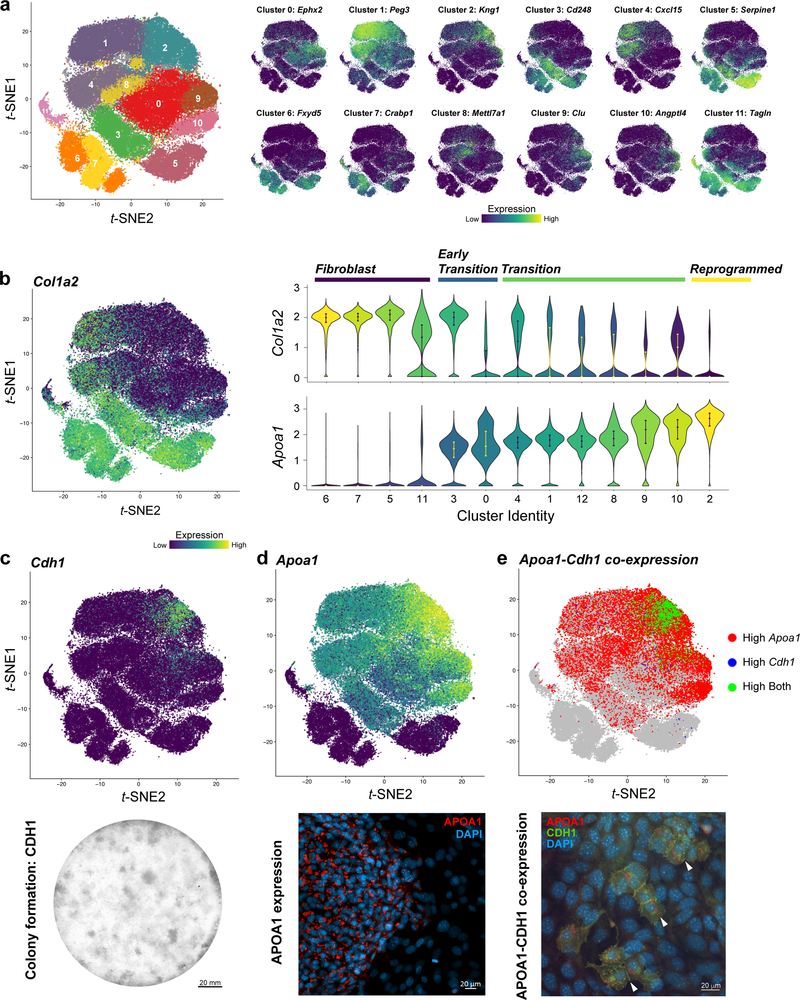
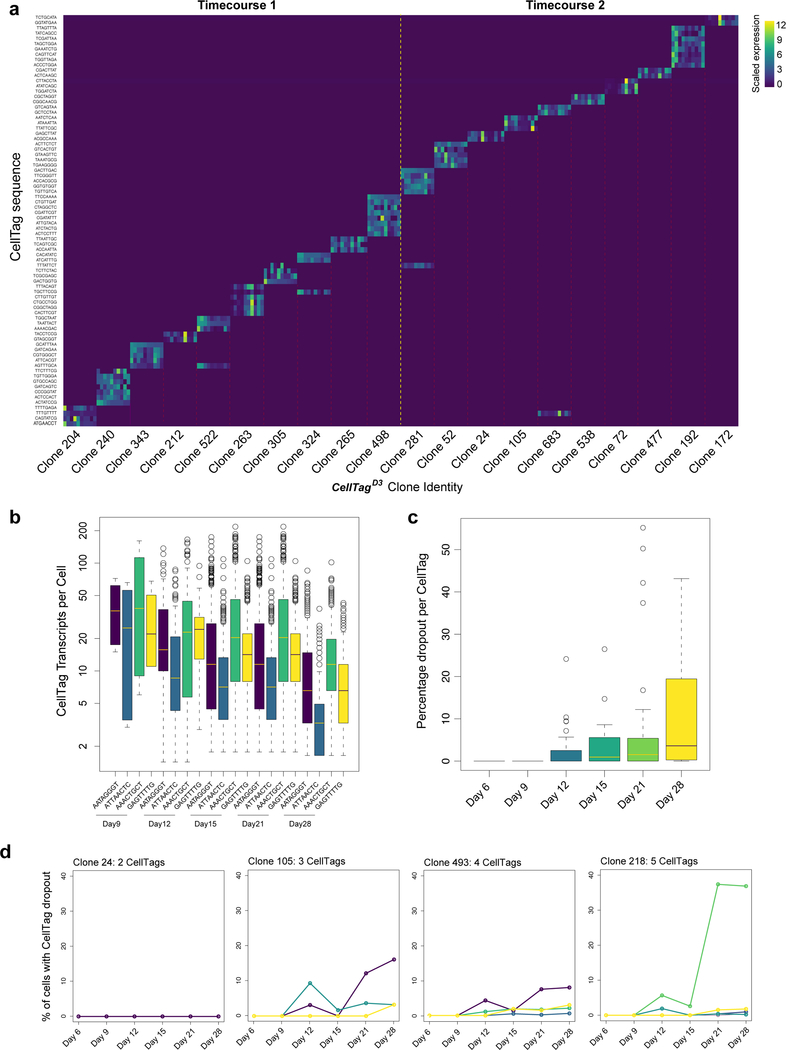
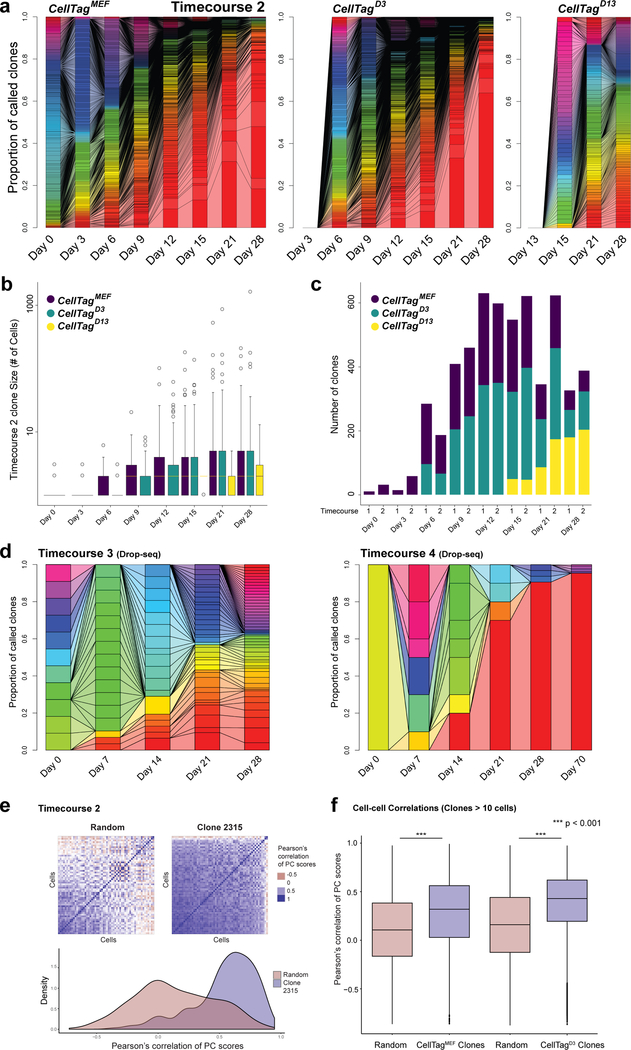
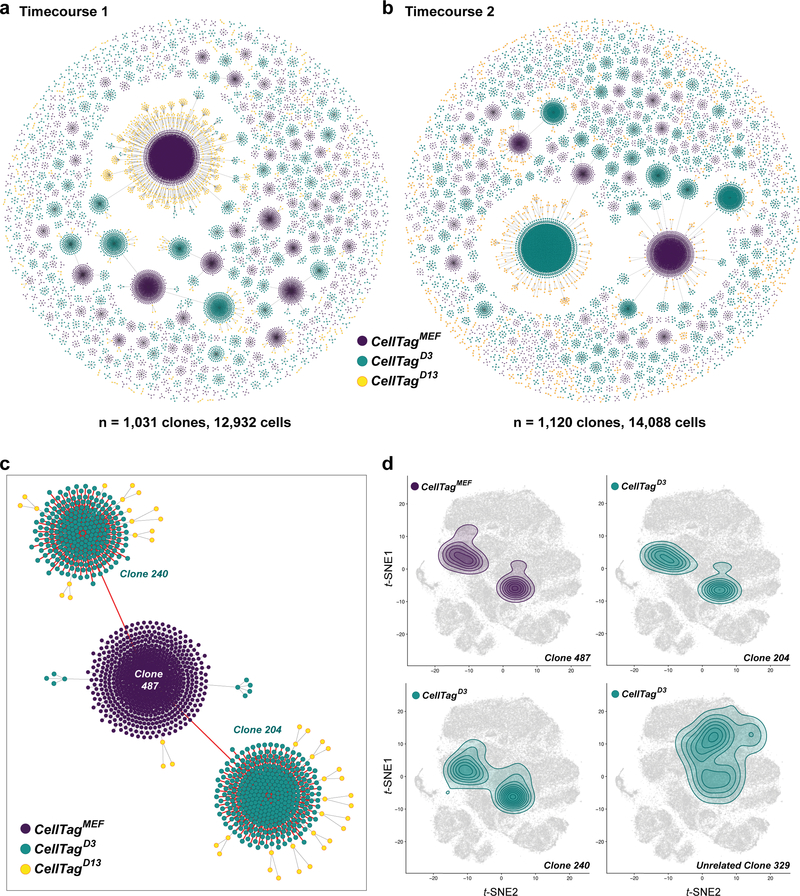
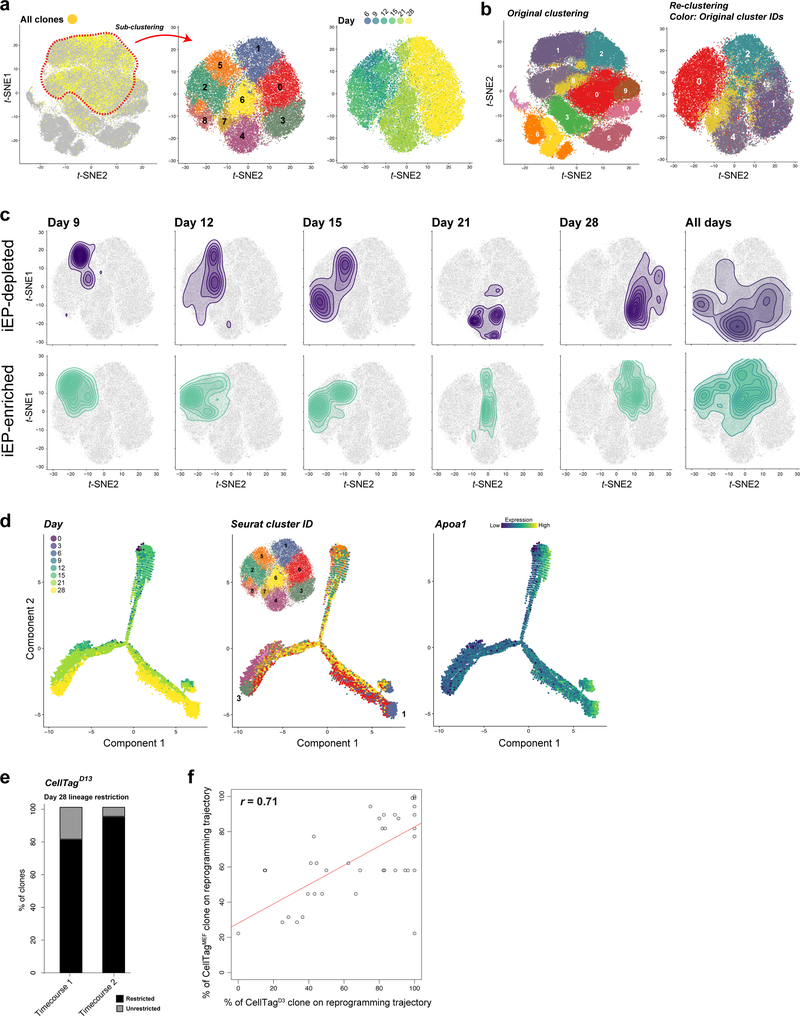
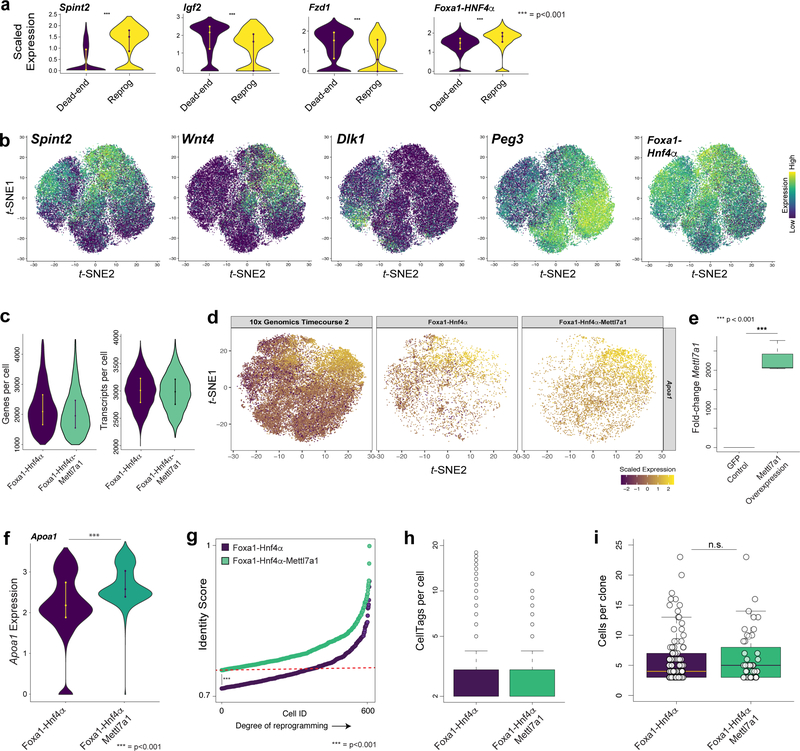
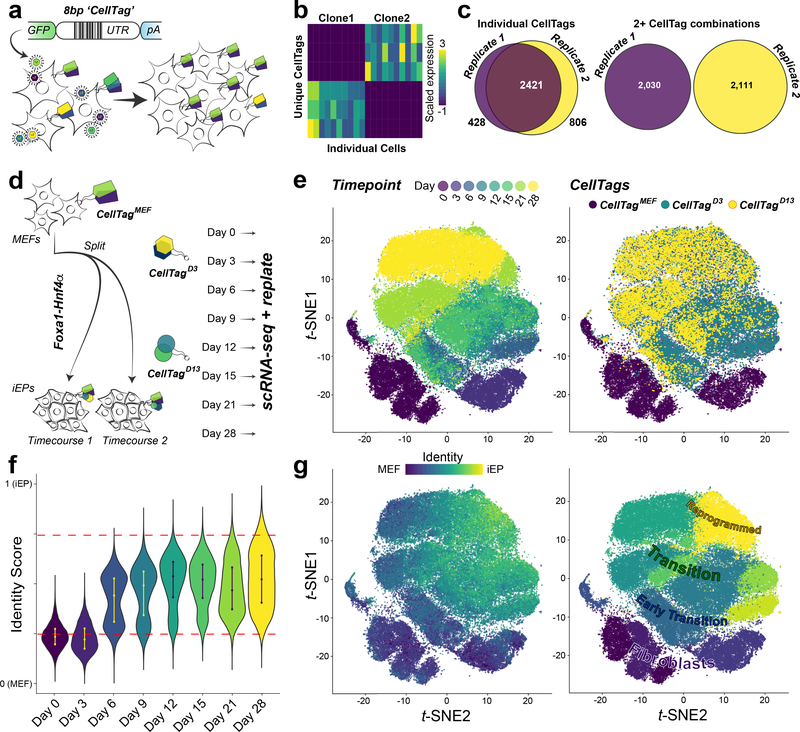
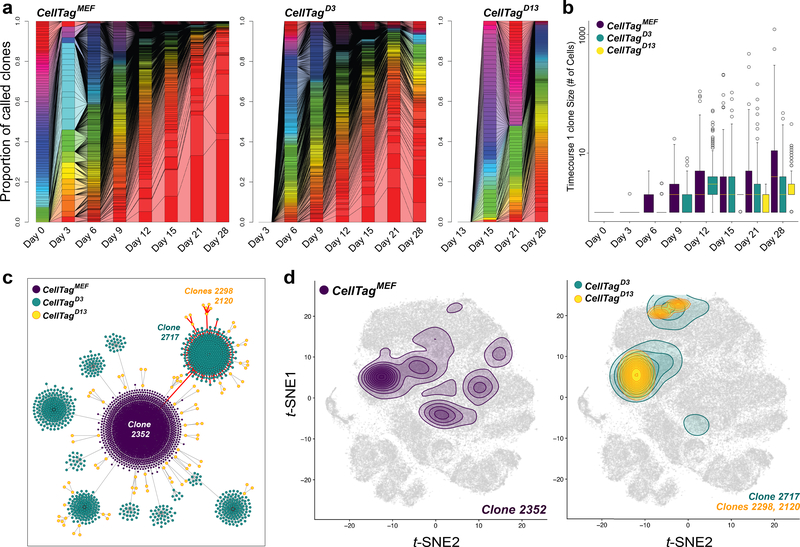
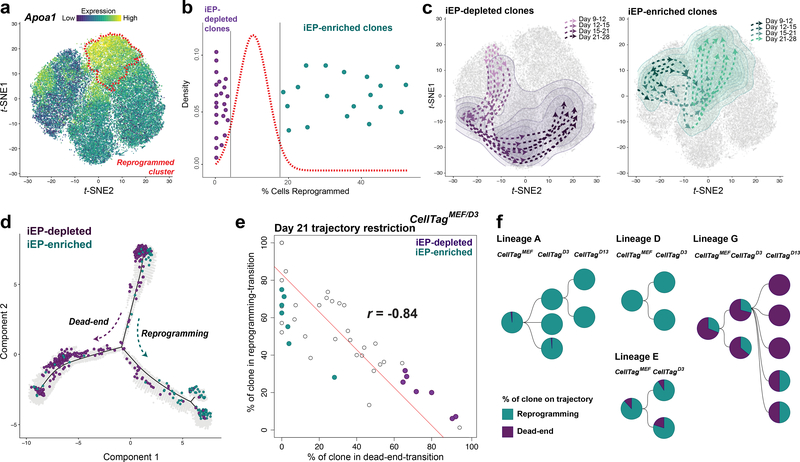
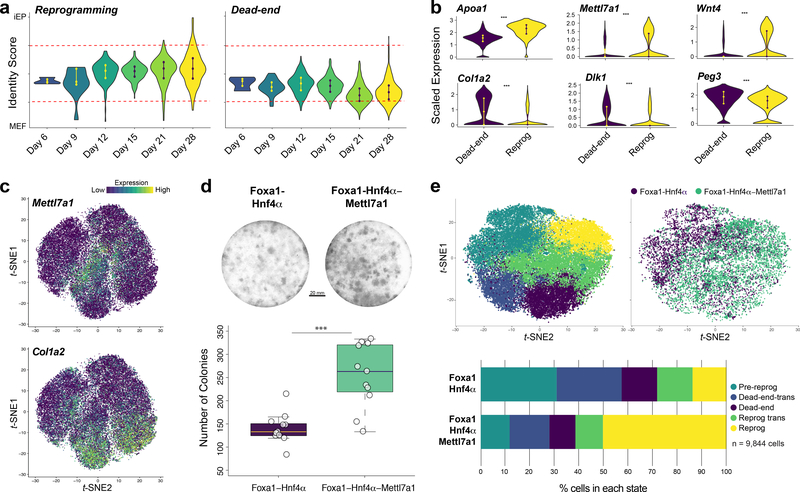
Direct lineage reprogramming involves the conversion of cellular identity. Single-cell technologies are useful for deconstructing the considerable heterogeneity that emerges during lineage conversion. However, lineage relationships are typically lost during cell processing, complicating trajectory reconstruction. Here we present 'CellTagging', a combinatorial cell-indexing methodology that enables parallel capture of clonal history and cell identity, in which sequential rounds of cell labelling enable the construction of multi-level lineage trees. CellTagging and longitudinal tracking of fibroblast to induced endoderm progenitor reprogramming reveals two distinct trajectories: one leading to successfully reprogrammed cells, and one leading to a 'dead-end' state, paths determined in the earliest stages of lineage conversion. We find that expression of a putative methyltransferase, Mettl7a1, is associated with the successful reprogramming trajectory; adding Mettl7a1 to the reprogramming cocktail increases the yield of induced endoderm progenitors. Together, these results demonstrate the utility of our lineage-tracing method for revealing the dynamics of direct reprogramming.
Figures
Comment in
-
Tagged reprogramming.Nat Methods. 2019 Feb;16(2):144. doi: 10.1038/s41592-019-0320-3. Nat Methods. 2019. PMID: 30700894 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
