

The plasma peptidome
- PMID: 30519149
- PMCID: PMC6271647
- DOI: 10.1186/s12014-018-9211-3
The plasma peptidome
Abstract
Background: It may be possible to discover new diagnostic or therapeutic peptides or proteins from blood plasma using LC-ESI-MS/MS to identify, with a linear quadrupole ion trap to identify, quantify and compare the statistical distributions of peptides cleaved ex vivo from plasma samples from different clinical populations.
Methods: A systematic method for the organic fractionation of plasma peptides was applied to identify and quantify the endogenous tryptic peptides from human plasma from multiple institutions by C18 HPLC followed nano electrospray ionization and tandem mass spectrometry (LC-ESI-MS/MS) with a linear quadrupole ion trap. The endogenous tryptic peptides, or tryptic phospho peptides (i.e. without exogenous digestion), were extracted in a mixture of organic solvent and water, dried and collected by preparative C18. The tryptic peptides from 6 institutions with 12 different disease and normal EDTA plasma populations, alongside ice cold controls for pre-analytical variation, were characterized by mass spectrometry. Each patient plasma was precipitated in 90% acetonitrile and the endogenous tryptic peptides extracted by a stepwise gradient of increasing water and then formic acid resulting in 10 sub-fractions. The fractionated peptides were manually collected over preparative C18 and injected for 1508 LC-ESI-MS/MS experiments analyzed in SQL Server R.
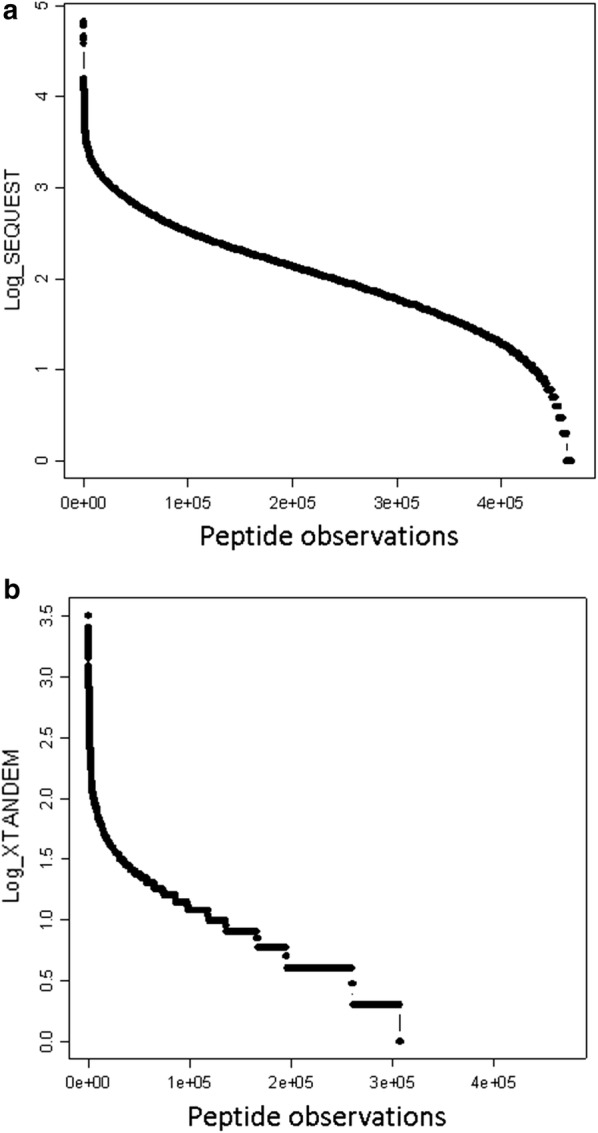
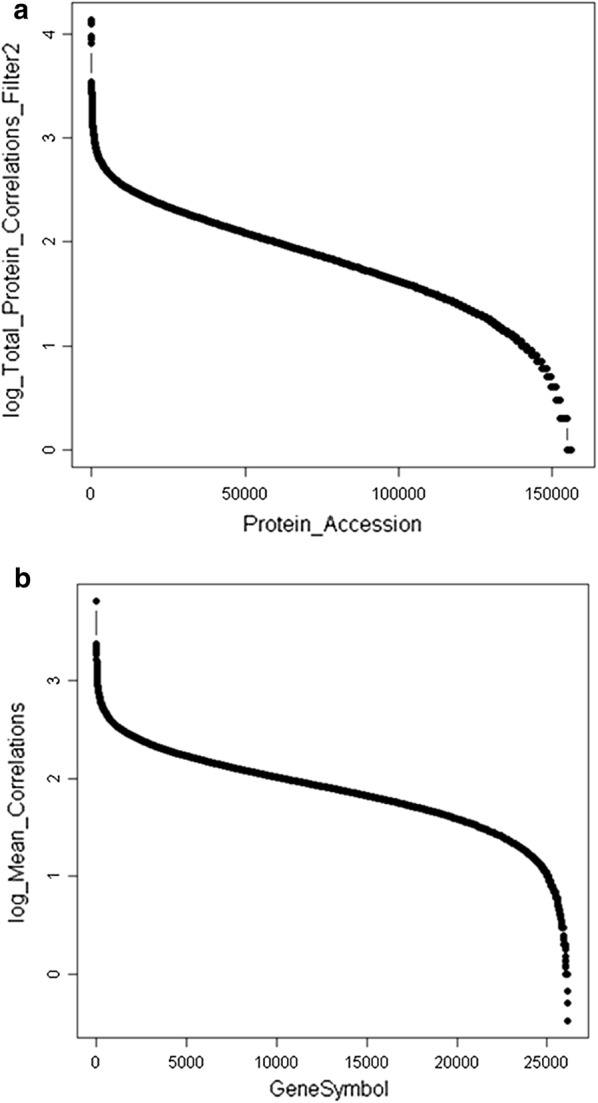
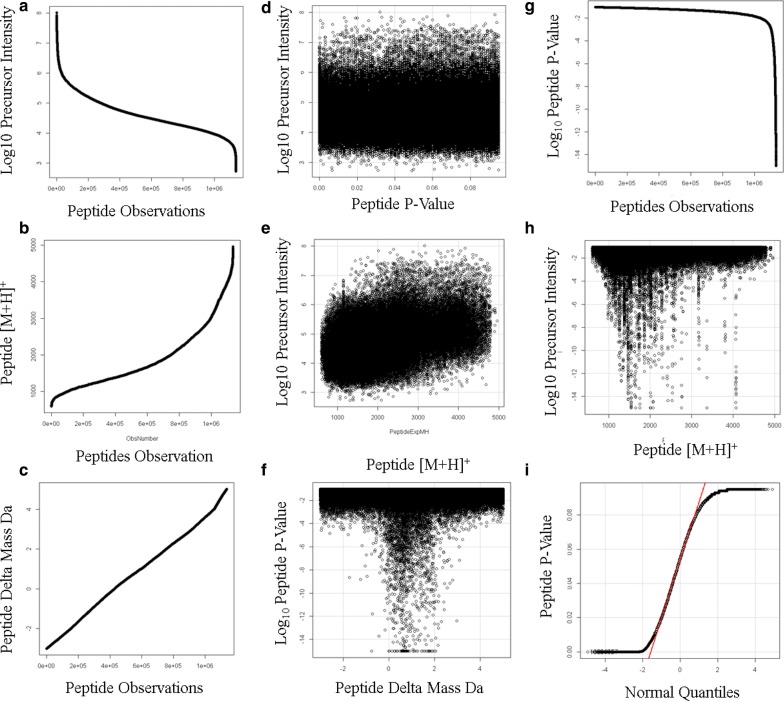
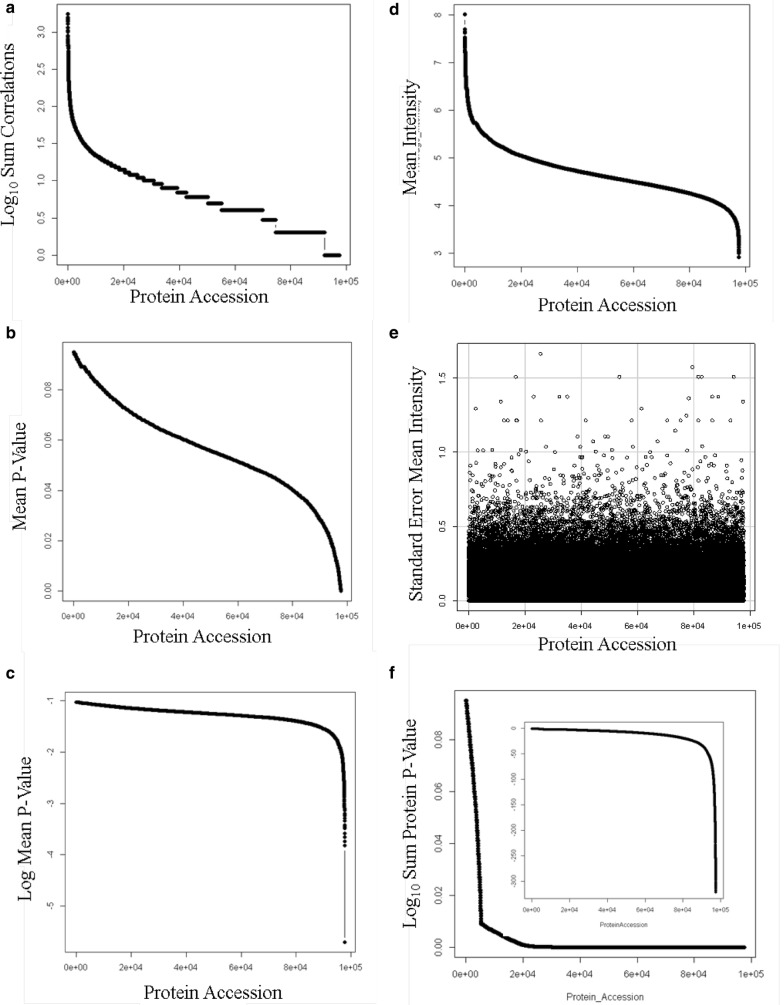
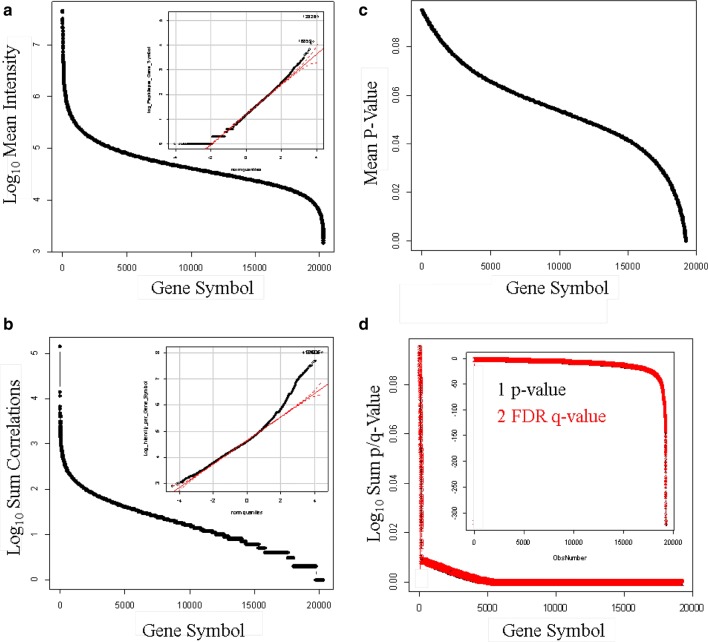
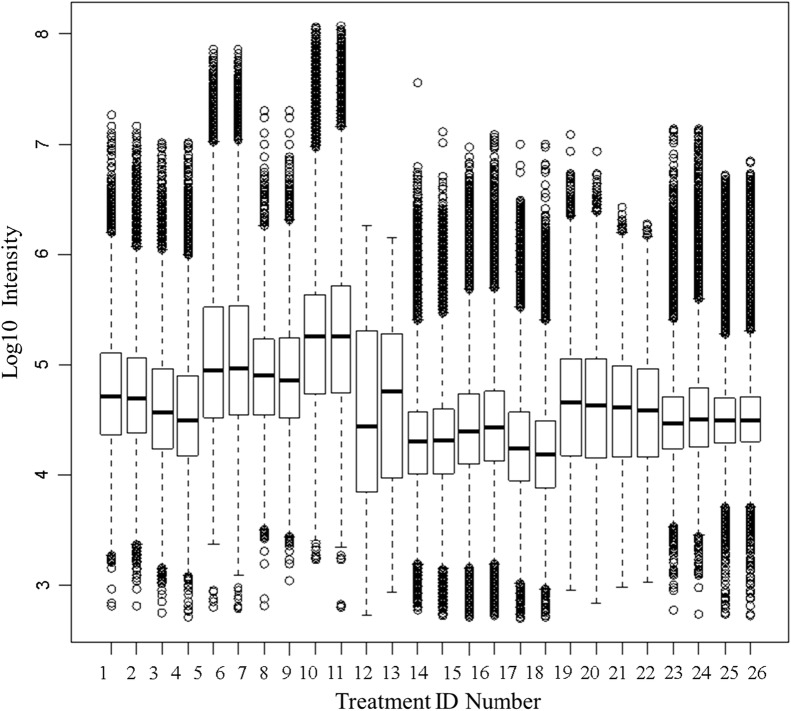
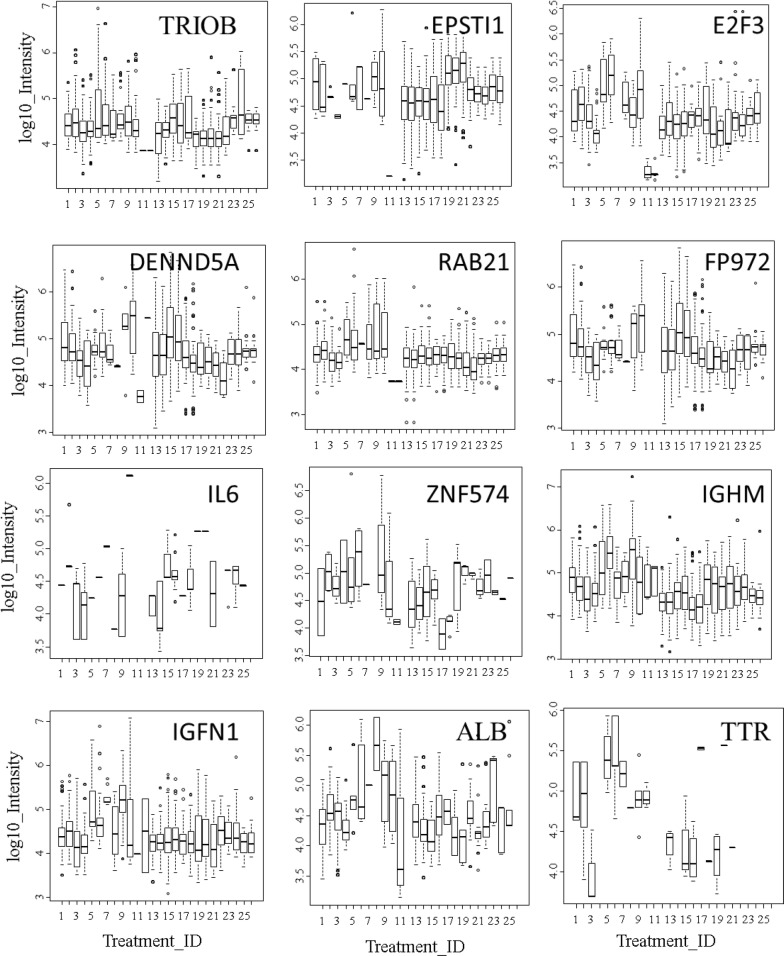
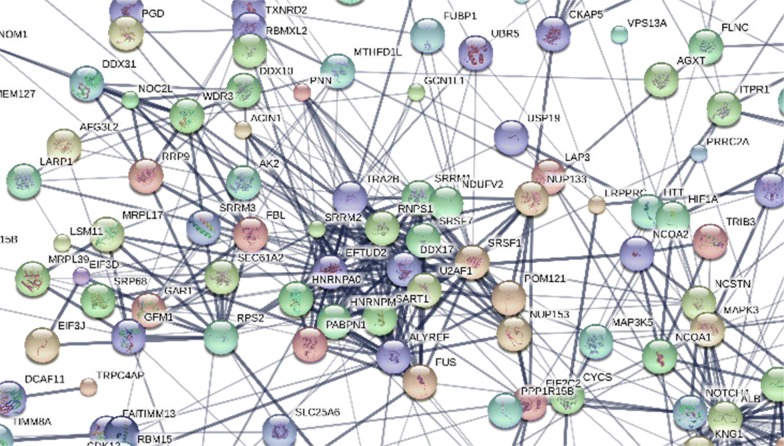
Results: Peptides that were cleaved in human plasma by a tryptic activity ex vivo provided convenient and sensitive access to most human proteins in plasma that show differences in the frequency or intensity of proteins observed across populations that may have clinical significance. Combination of step wise organic extraction of 200 μL of plasma with nano electrospray resulted in the confident identification and quantification ~ 14,000 gene symbols by X!TANDEM that is the largest number of blood proteins identified to date and shows that you can monitor the ex vivo proteolysis of most human proteins, including interleukins, from blood. A total of 15,968,550 MS/MS spectra ≥ E4 intensity counts were correlated by the SEQUEST and X!TANDEM algorithms to a federated library of 157,478 protein sequences that were filtered for best charge state (2+ or 3+) and peptide sequence in SQL Server resulting in 1,916,672 distinct best-fit peptide correlations for analysis with the R statistical system. SEQUEST identified some 140,054 protein accessions, or some ~ 26,000 gene symbols, proteins or loci, with at least 5 independent correlations. The X!TANDEM algorithm made at least 5 best fit correlations to more than 14,000 protein gene symbols with p-values and FDR corrected q-values of ~ 0.001 or less. Log10 peptide intensity values showed a Gaussian distribution from E8 to E4 arbitrary counts by quantile plot, and significant variation in average precursor intensity across the disease and controls treatments by ANOVA with means compared by the Tukey-Kramer test. STRING analysis of the top 2000 gene symbols showed a tight association of cellular proteins that were apparently present in the plasma as protein complexes with related cellular components, molecular functions and biological processes.
Conclusions: The random and independent sampling of pre-fractionated blood peptides by LC-ESI-MS/MS with SQL Server-R analysis revealed the largest plasma proteome to date and was a practical method to quantify and compare the frequency or log10 intensity of individual proteins cleaved ex vivo across populations of plasma samples from multiple clinical locations to discover treatment-specific variation using classical statistics suitable for clinical science. It was possible to identify and quantify nearly all human proteins from EDTA plasma and compare the results of thousands of LC-ESI-MS/MS experiments from multiple clinical populations using standard database methods in SQL Server and classical statistical strategies in the R data analysis system.
Keywords: Electrospray ionization tandem mass spectrometry; Endogenous tryptic peptides phospho peptides; Human EDTA plasma; LC–ESI–MS/MS; Linear quadrupole ion trap; Nano chromatography; Organic extraction.
Figures
References
-
- Putnam F. The plasma proteins: structure function, and genetic control. 2. New York: Academic Press; 1975.
-
- Burtis CA, Ashwood ER, DE Bruns, editors. Tietz fundamentals of clinical chemistry. Philadelphia: Saunders; 2001. p. 1091.
LinkOut - more resources
Full Text Sources