

Cellulose-specific Type B carbohydrate binding modules: understanding oligomeric and non-crystalline substrate recognition mechanisms
- PMID: 30519283
- PMCID: PMC6267901
- DOI: 10.1186/s13068-018-1321-7
Cellulose-specific Type B carbohydrate binding modules: understanding oligomeric and non-crystalline substrate recognition mechanisms
Abstract
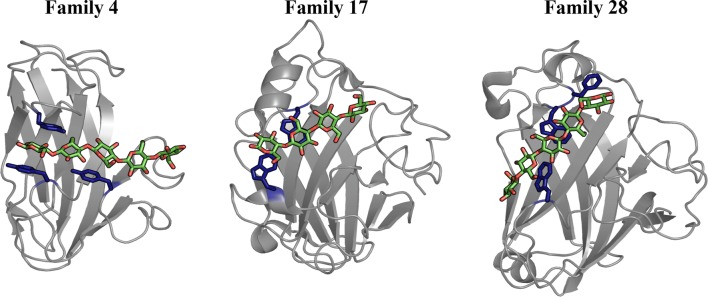
Background: Effective enzymatic degradation of crystalline polysaccharides requires a synergistic cocktail of hydrolytic enzymes tailored to the wide-ranging degree of substrate crystallinity. To accomplish this type of targeted carbohydrate recognition, nature produces multi-modular enzymes, having at least one catalytic domain appended to one or more carbohydrate binding modules (CBMs). The Type B CBM categorization encompasses several families (i.e., protein folds) of CBMs that are generally thought to selectively bind oligomeric polysaccharides; however, a subset of cellulose-specific CBM families (17 and 28) appear to bind non-crystalline cellulose more tightly than oligomers and in a manner that discriminates between surface topology.
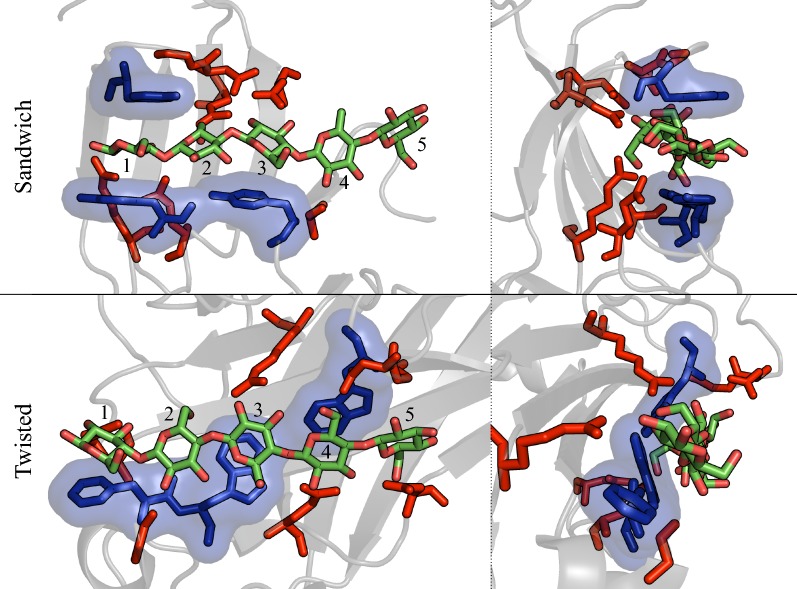
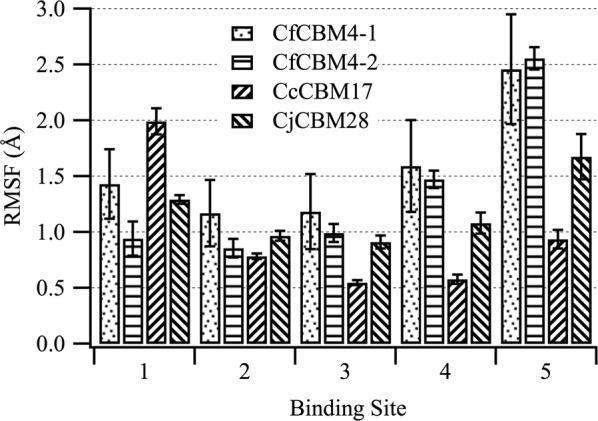
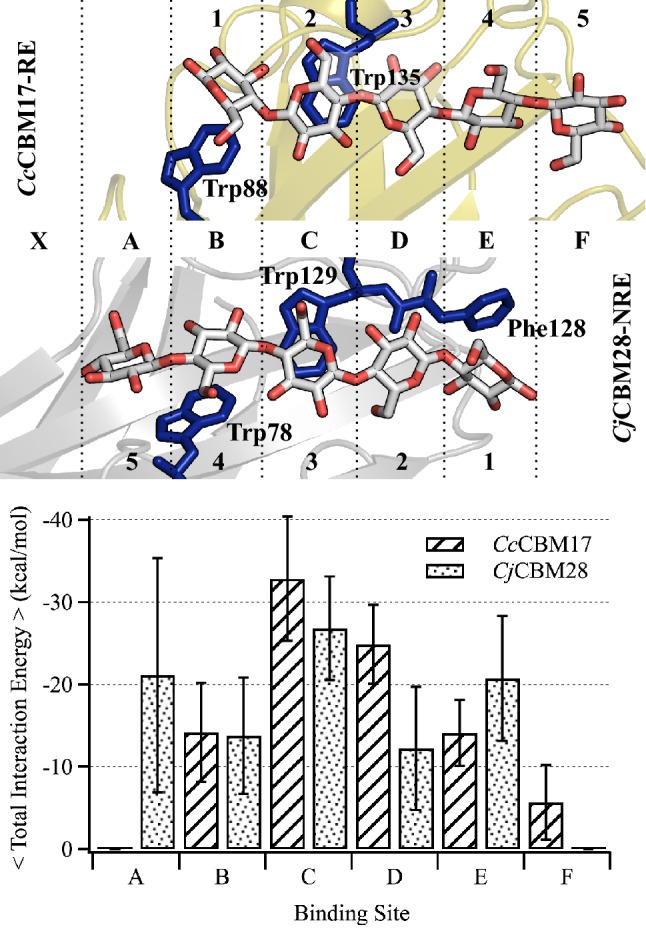
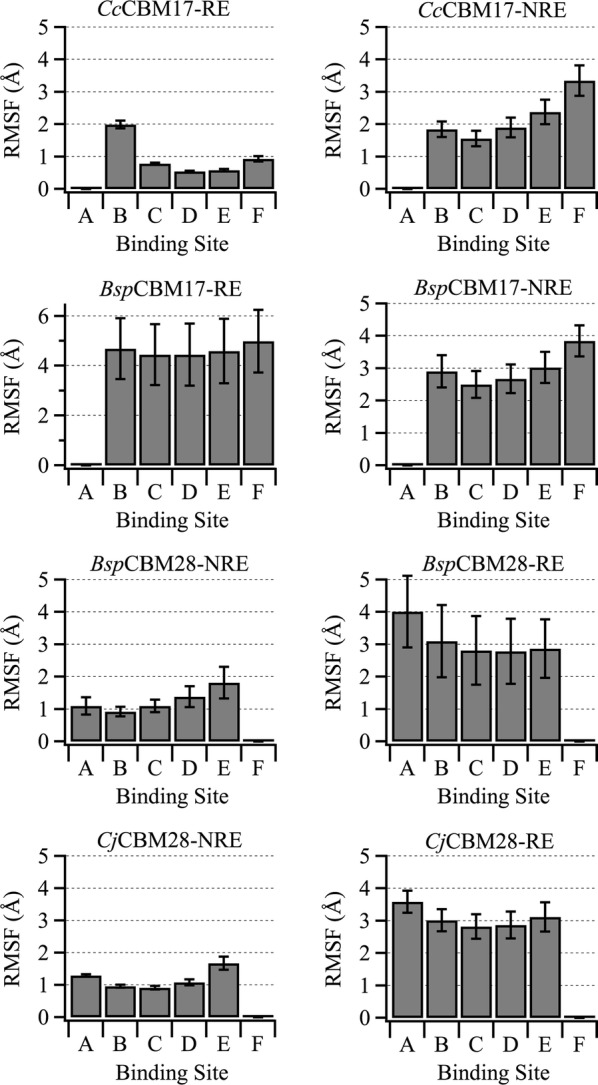
Results: To provide insight into this unexplained phenomenon, we investigated the molecular-level origins of oligomeric and non-crystalline carbohydrate recognition in cellulose-specific Type B CBMs using molecular dynamics (MD) simulation and free energy calculations. Examining two CBMs from three different families (4, 17, and 28), we describe how protein-ligand dynamics contribute to observed variations in binding affinity of oligomers within the same CBM family. Comparisons across the three CBM families identified factors leading to modified functionality prohibiting competitive binding, despite similarity in sequence and specificity. Using free energy perturbation with Hamiltonian replica exchange MD, we also examined the hypothesis that the open topology of the binding grooves in families 17 and 28 necessitates tight binding of an oligomer, while the more confined family 4 binding groove does not require the same degree of tight binding. Finally, we elucidated the mechanisms of non-crystalline carbohydrate recognition by modeling CBMs complexed with a partially decrystallized cellulose substrate. Molecular simulation provided structural and dynamic data for direct comparison to oligomeric modes of carbohydrate recognition, and umbrella sampling MD was used to determine ligand binding free energy. Comparing both protein-carbohydrate interactions and ligand binding free energies, which were in good agreement with experimental values, we confirmed the hypothesis that family 17 and 28 CBMs bind non-crystalline cellulose and oligomers with different affinities (i.e., high and low).
Conclusions: Our study provides an unprecedented level of insight into the complex solid and soluble carbohydrate substrate recognition mechanisms of Type B CBMs, the findings of which hold considerable promise for enhancing lignocellulosic biomass conversion technology and development of plant cell wall probes.
Keywords: Amorphous cellulose; Carbohydrate–aromatic stacking; Free energy perturbation; Multi-modular glycoside hydrolases; β-Sandwich fold.
Figures




References
LinkOut - more resources
Full Text Sources
