

PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity
- PMID: 30523025
- PMCID: PMC6385748
- DOI: 10.2337/db18-0363
PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity
Abstract
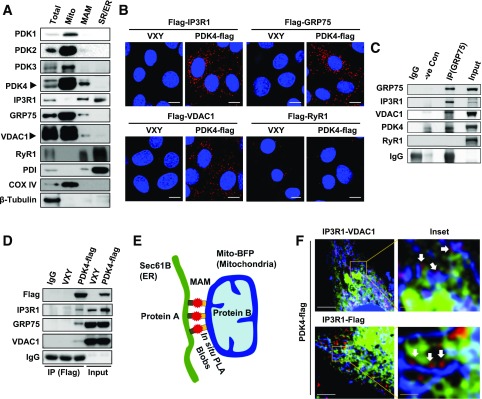
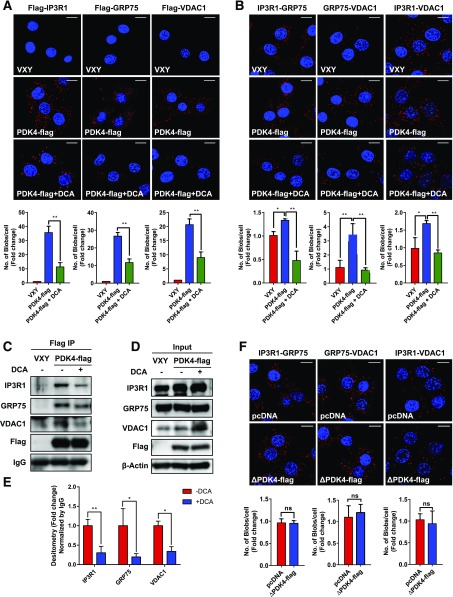
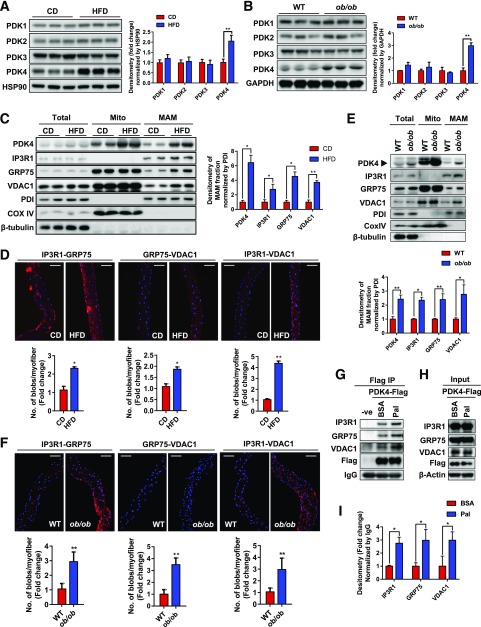
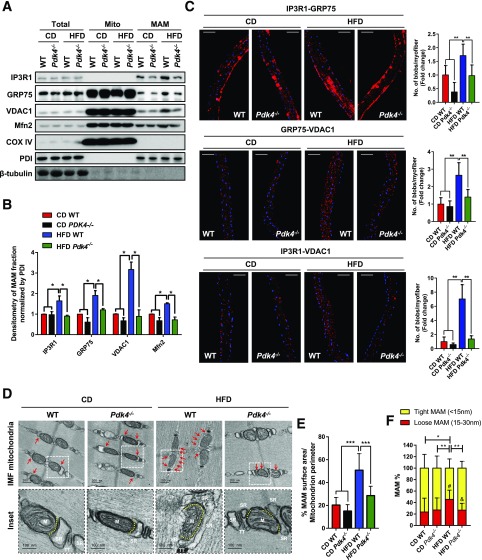
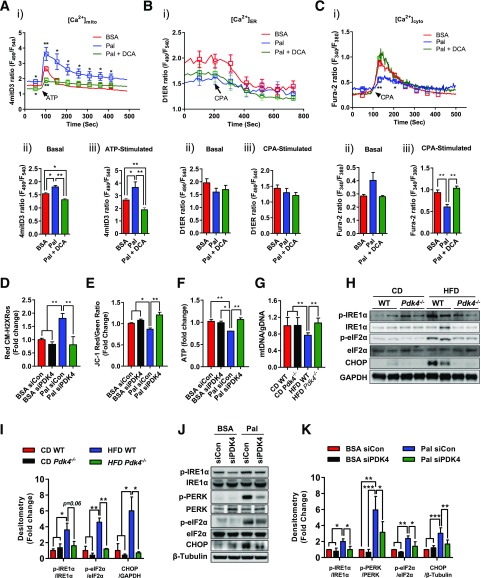
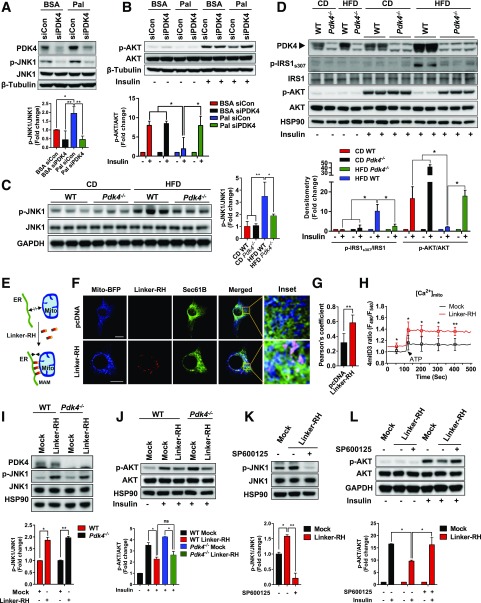
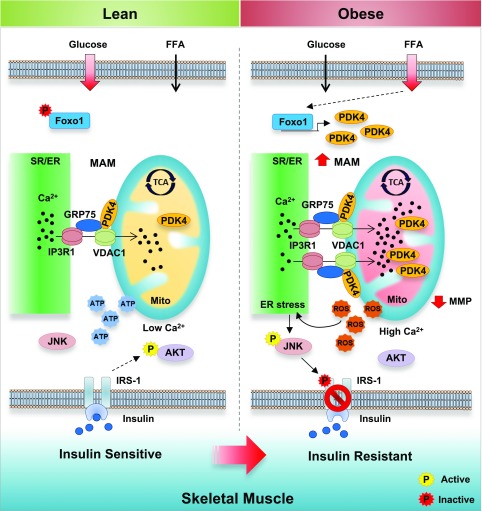
Mitochondria-associated endoplasmic reticulum membrane (MAM) is a structural link between mitochondria and endoplasmic reticulum (ER). MAM regulates Ca2+ transport from the ER to mitochondria via an IP3R1-GRP75-VDAC1 complex-dependent mechanism. Excessive MAM formation may cause mitochondrial Ca2+ overload and mitochondrial dysfunction. However, the exact implication of MAM formation in metabolic syndromes remains debatable. Here, we demonstrate that PDK4 interacts with and stabilizes the IP3R1-GRP75-VDAC1 complex at the MAM interface. Obesity-induced increase in PDK4 activity augments MAM formation and suppresses insulin signaling. Conversely, PDK4 inhibition dampens MAM formation and improves insulin signaling by preventing MAM-induced mitochondrial Ca2+ accumulation, mitochondrial dysfunction, and ER stress. Furthermore, Pdk4-/- mice exhibit reduced MAM formation and are protected against diet-induced skeletal muscle insulin resistance. Finally, forced formation and stabilization of MAMs with synthetic ER-mitochondria linker prevented the beneficial effects of PDK4 deficiency on insulin signaling. Overall, our findings demonstrate a critical mediatory role of PDK4 in the development of skeletal muscle insulin resistance via enhancement of MAM formation.
© 2018 by the American Diabetes Association.
Figures
References
-
- Tubbs E, Theurey P, Vial G, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014;63:3279–3294 - PubMed
-
- Rieusset J, Fauconnier J, Paillard M, et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016;59:614–623 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
