

Polycystic kidney disease
- PMID: 30523303
- PMCID: PMC6592047
- DOI: 10.1038/s41572-018-0047-y
Polycystic kidney disease
Abstract
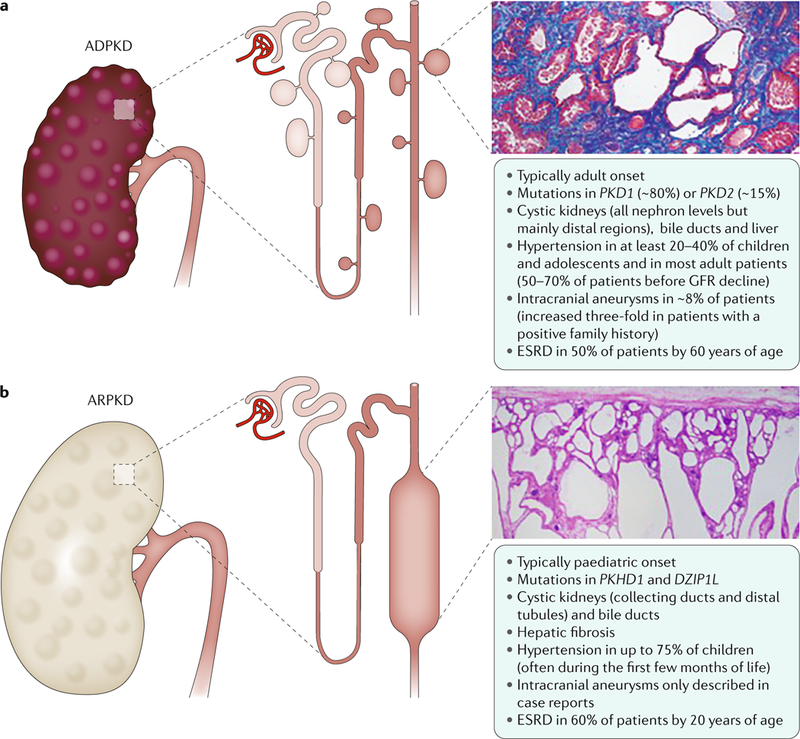
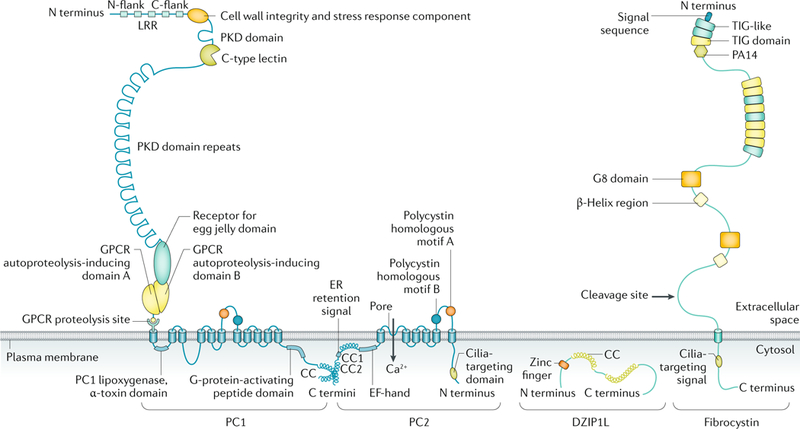
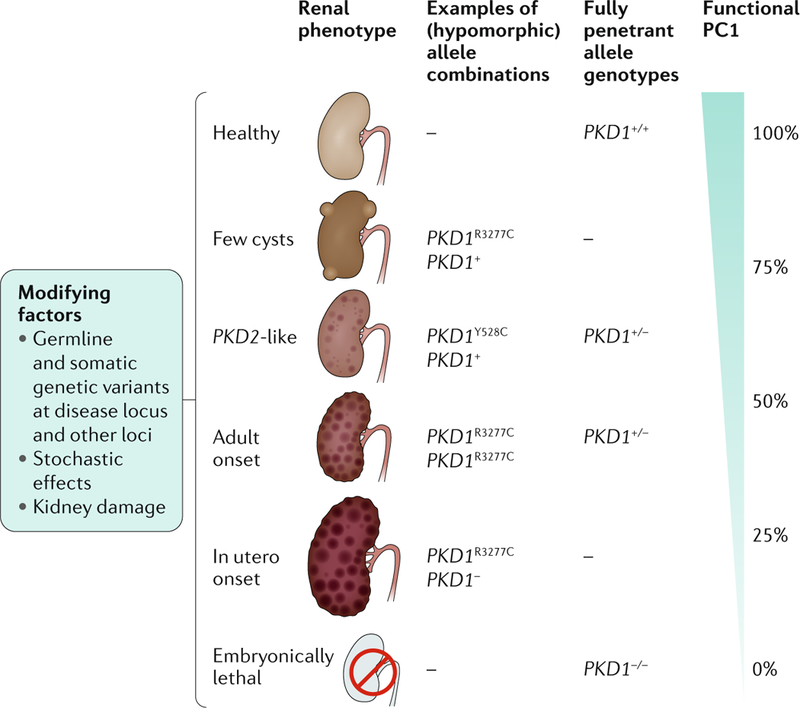
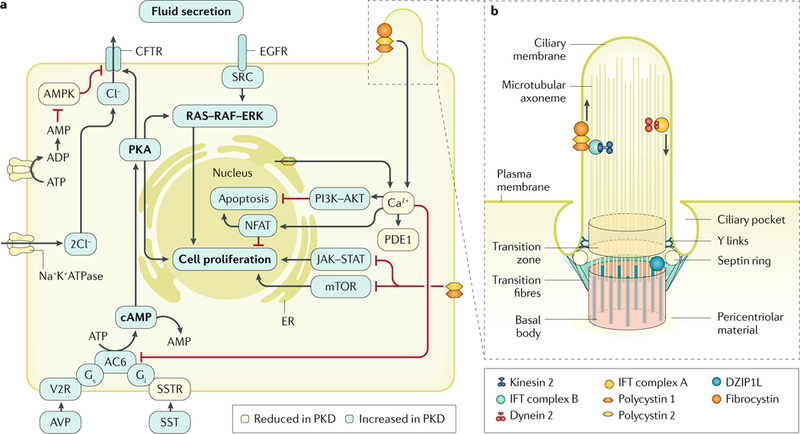
Cystic kidneys are common causes of end-stage renal disease, both in children and in adults. Autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) are cilia-related disorders and the two main forms of monogenic cystic kidney diseases. ADPKD is a common disease that mostly presents in adults, whereas ARPKD is a rarer and often more severe form of polycystic kidney disease (PKD) that usually presents perinatally or in early childhood. Cell biological and clinical research approaches have expanded our knowledge of the pathogenesis of ADPKD and ARPKD and revealed some mechanistic overlap between them. A reduced 'dosage' of PKD proteins is thought to disturb cell homeostasis and converging signalling pathways, such as Ca2+, cAMP, mechanistic target of rapamycin, WNT, vascular endothelial growth factor and Hippo signalling, and could explain the more severe clinical course in some patients with PKD. Genetic diagnosis might benefit families and improve the clinical management of patients, which might be enhanced even further with emerging therapeutic options. However, many important questions about the pathogenesis of PKD remain. In this Primer, we provide an overview of the current knowledge of PKD and its treatment.
Figures
References
-
- Torres VE, Harris PC & Pirson Y Autosomal dominant polycystic kidney disease. Lancet 369, 1287–1301 (2007). - PubMed
-
- Bergmann C & Weiskirchen R It’s not all in the cilium, but on the road to it: genetic interaction network in polycystic kidney and liver diseases and how trafficking and quality control matter. J. Hepatol.56, 1201–1203 (2012). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
