

Aberrant Inclusion of a Poison Exon Causes Dravet Syndrome and Related SCN1A-Associated Genetic Epilepsies
- PMID: 30526861
- PMCID: PMC6288405
- DOI: 10.1016/j.ajhg.2018.10.023
Aberrant Inclusion of a Poison Exon Causes Dravet Syndrome and Related SCN1A-Associated Genetic Epilepsies
Abstract
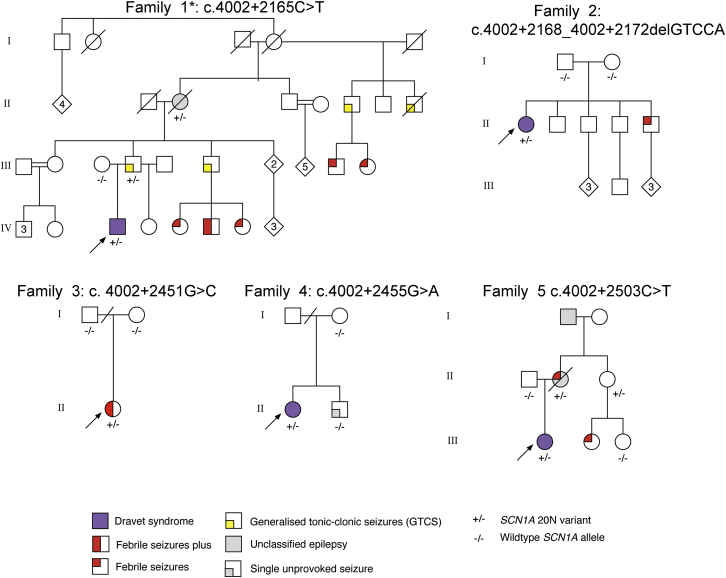
Developmental and epileptic encephalopathies (DEEs) are a group of severe epilepsies characterized by refractory seizures and developmental impairment. Sequencing approaches have identified causal genetic variants in only about 50% of individuals with DEEs.1-3 This suggests that unknown genetic etiologies exist, potentially in the ∼98% of human genomes not covered by exome sequencing (ES). Here we describe seven likely pathogenic variants in regions outside of the annotated coding exons of the most frequently implicated epilepsy gene, SCN1A, encoding the alpha-1 sodium channel subunit. We provide evidence that five of these variants promote inclusion of a "poison" exon that leads to reduced amounts of full-length SCN1A protein. This mechanism is likely to be broadly relevant to human disease; transcriptome studies have revealed hundreds of poison exons,4,5 including some present within genes encoding other sodium channels and in genes involved in neurodevelopment more broadly.6 Future research on the mechanisms that govern neuronal-specific splicing behavior might allow researchers to co-opt this system for RNA therapeutics.
Keywords: Dravet syndrome; SCN1A; alternative splicing; epilepsy; genome sequencing; noncoding; poison exon; variant interpretation.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Zhang Q., Li J., Zhao Y., Bao X., Wei L., Wang J. Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin Genet. 2016;91:717–724. - PubMed
-
- Howell K.B., Eggers S., Dalziel K., Riseley J., Mandelstam S., Myers C.T., McMahon J.M., Schneider A., Carvill G.L., Mefford H.C., Victorian Severe Epilepsy of Infancy Study Group A population-based cost-effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia. 2018;59:1177–1187. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
