

Experimental accuracy in protein structure refinement via molecular dynamics simulations
- PMID: 30530696
- PMCID: PMC6310835
- DOI: 10.1073/pnas.1811364115
Experimental accuracy in protein structure refinement via molecular dynamics simulations
Abstract
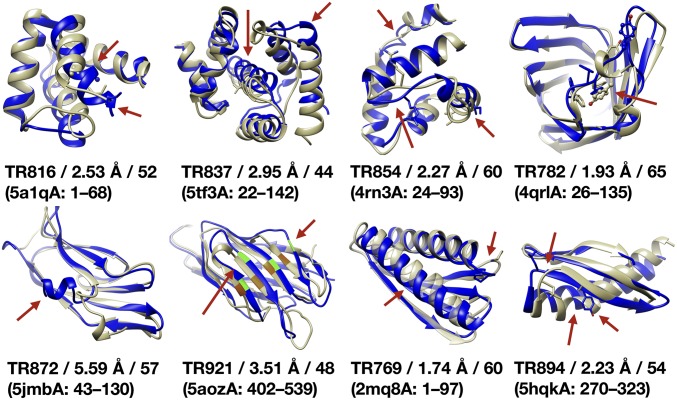
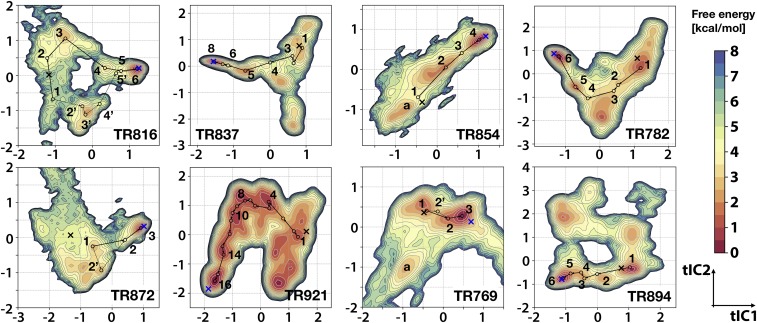
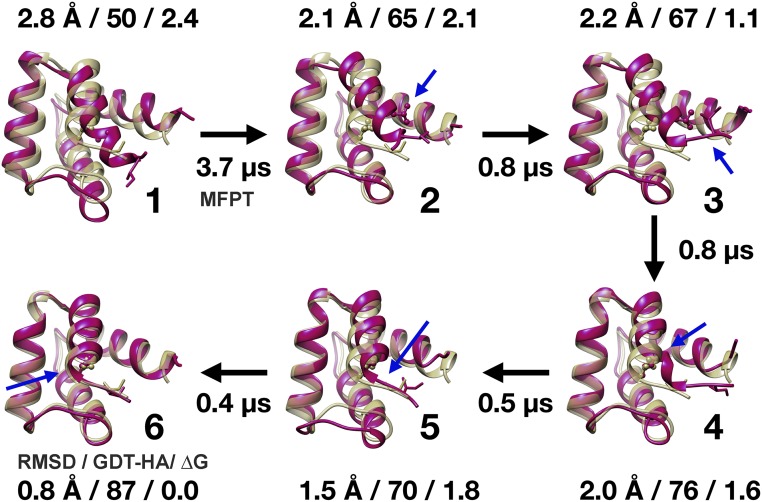
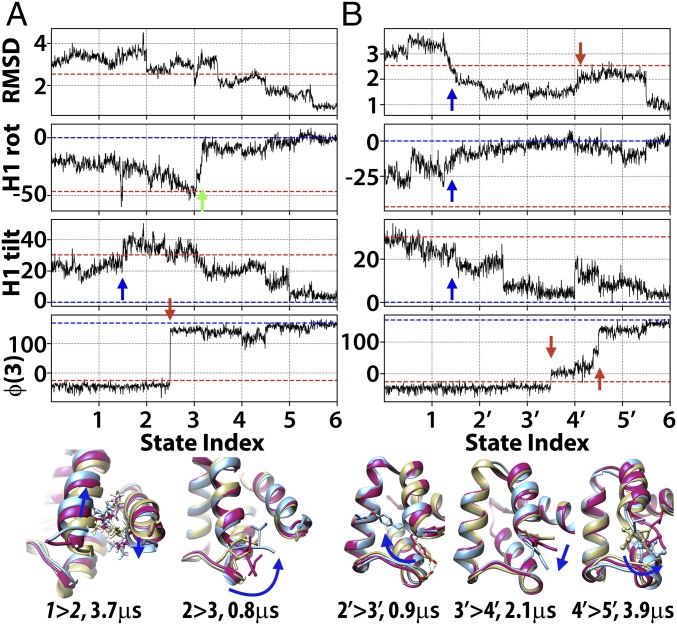
Refinement is the last step in protein structure prediction pipelines to convert approximate homology models to experimental accuracy. Protocols based on molecular dynamics (MD) simulations have shown promise, but current methods are limited to moderate levels of consistent refinement. To explore the energy landscape between homology models and native structures and analyze the challenges of MD-based refinement, eight test cases were studied via extensive simulations followed by Markov state modeling. In all cases, native states were found very close to the experimental structures and at the lowest free energies, but refinement was hindered by a rough energy landscape. Transitions from the homology model to the native states require the crossing of significant kinetic barriers on at least microsecond time scales. A significant energetic driving force toward the native state was lacking until its immediate vicinity, and there was significant sampling of off-pathway states competing for productive refinement. The role of recent force field improvements is discussed and transition paths are analyzed in detail to inform which key transitions have to be overcome to achieve successful refinement.
Keywords: Markov state model; conformational sampling; energy landscape; force field; protein structure prediction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
