

A map of constrained coding regions in the human genome
- PMID: 30531870
- PMCID: PMC6589356
- DOI: 10.1038/s41588-018-0294-6
A map of constrained coding regions in the human genome
Abstract
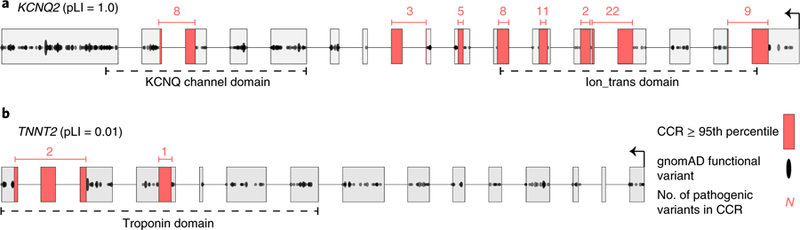
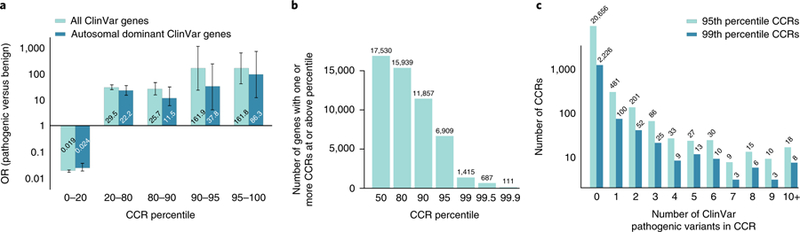
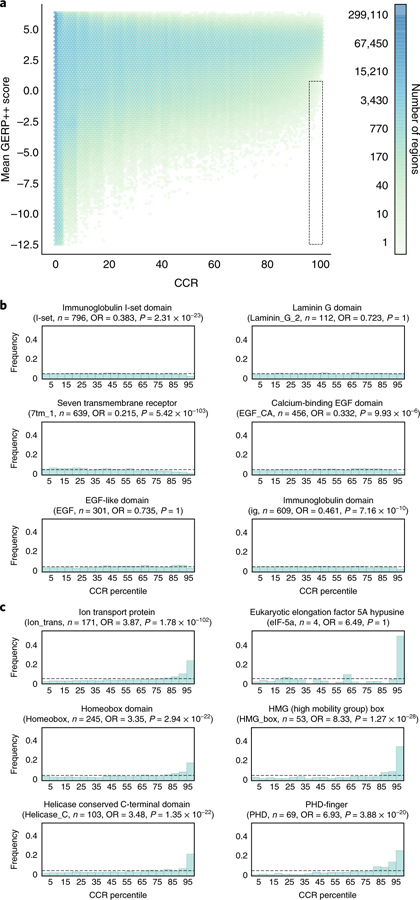
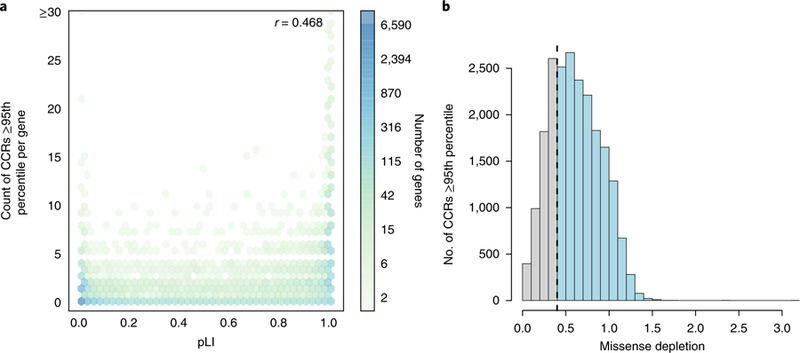
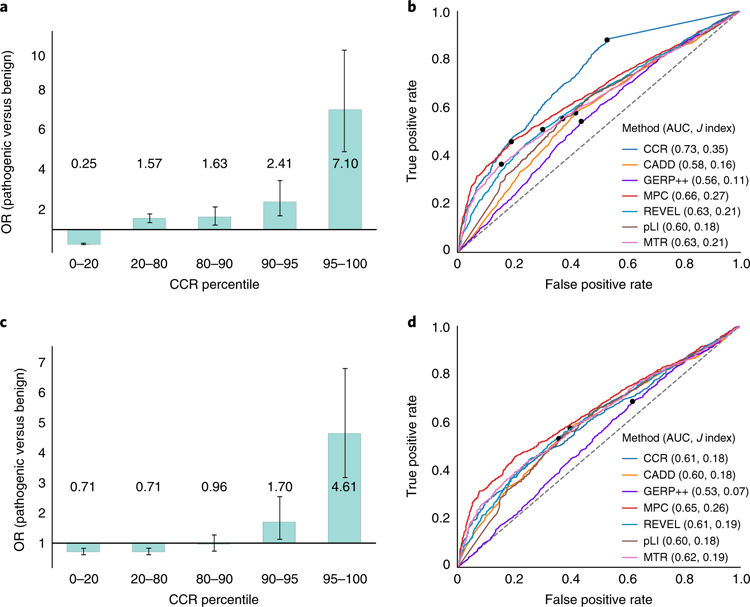
Deep catalogs of genetic variation from thousands of humans enable the detection of intraspecies constraint by identifying coding regions with a scarcity of variation. While existing techniques summarize constraint for entire genes, single gene-wide metrics conceal regional constraint variability within each gene. Therefore, we have created a detailed map of constrained coding regions (CCRs) by leveraging variation observed among 123,136 humans from the Genome Aggregation Database. The most constrained CCRs are enriched for pathogenic variants in ClinVar and mutations underlying developmental disorders. CCRs highlight protein domain families under high constraint and suggest unannotated or incomplete protein domains. The highest-percentile CCRs complement existing variant prioritization methods when evaluating de novo mutations in studies of autosomal dominant disease. Finally, we identify highly constrained CCRs within genes lacking known disease associations. This observation suggests that CCRs may identify regions under strong purifying selection that, when mutated, cause severe developmental phenotypes or embryonic lethality.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Wallis WA The statistical research group, 1942–1945. J. Am. Stat. Assoc 75, 320–330 (1980).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
