

The molecular landscape of glioma in patients with Neurofibromatosis 1
- PMID: 30531922
- PMCID: PMC6857804
- DOI: 10.1038/s41591-018-0263-8
The molecular landscape of glioma in patients with Neurofibromatosis 1
Abstract
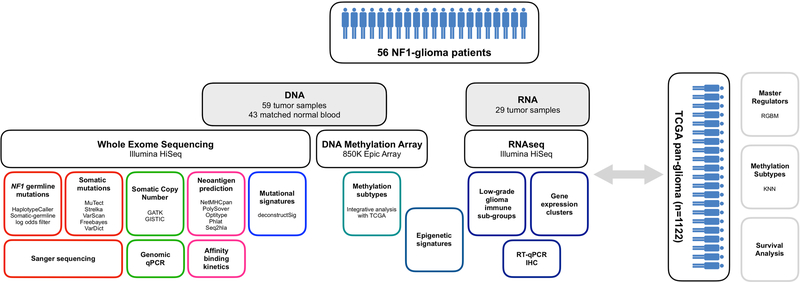
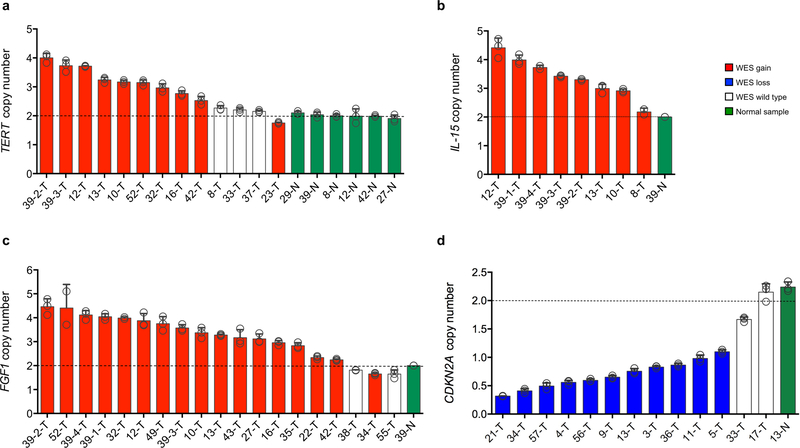
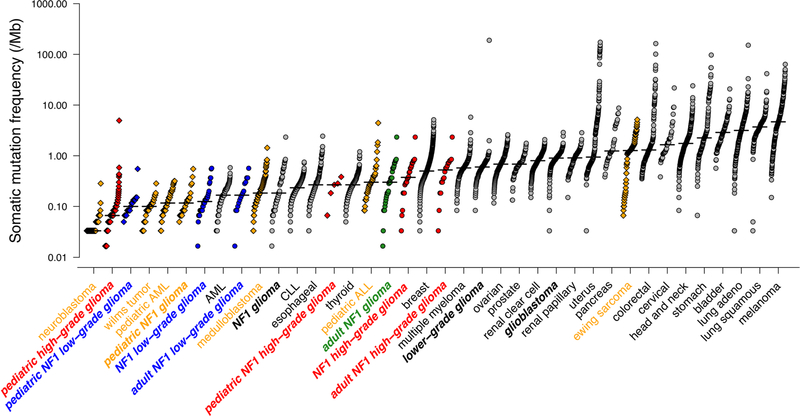
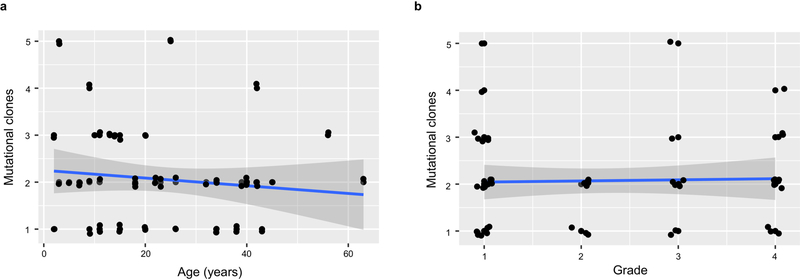
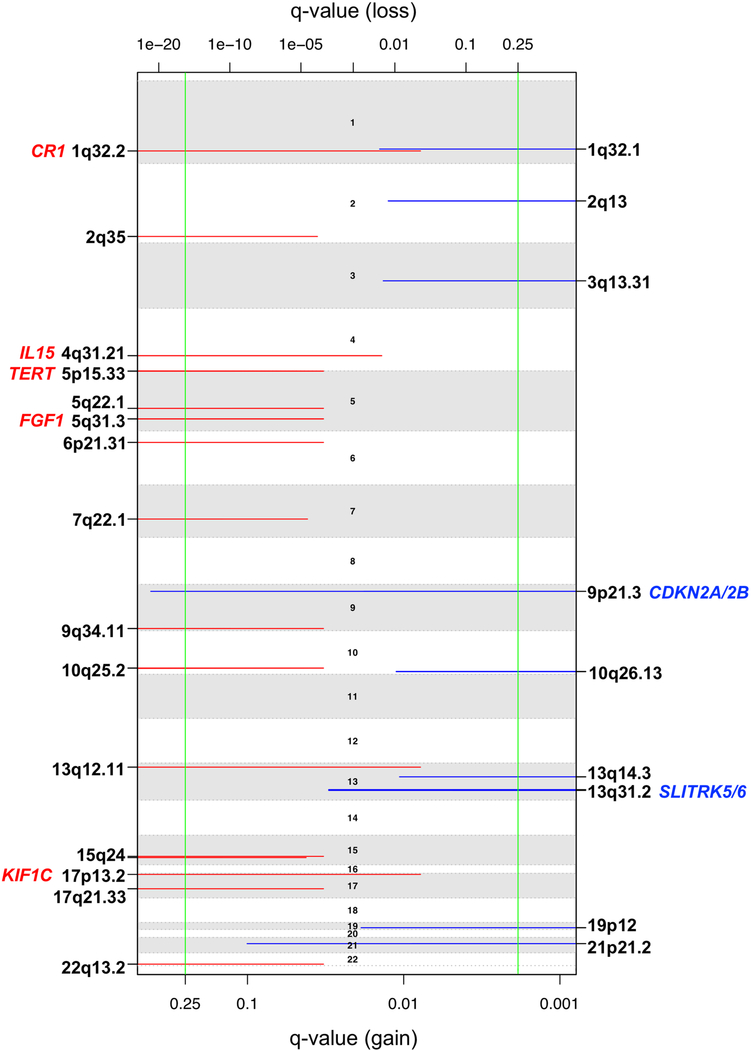
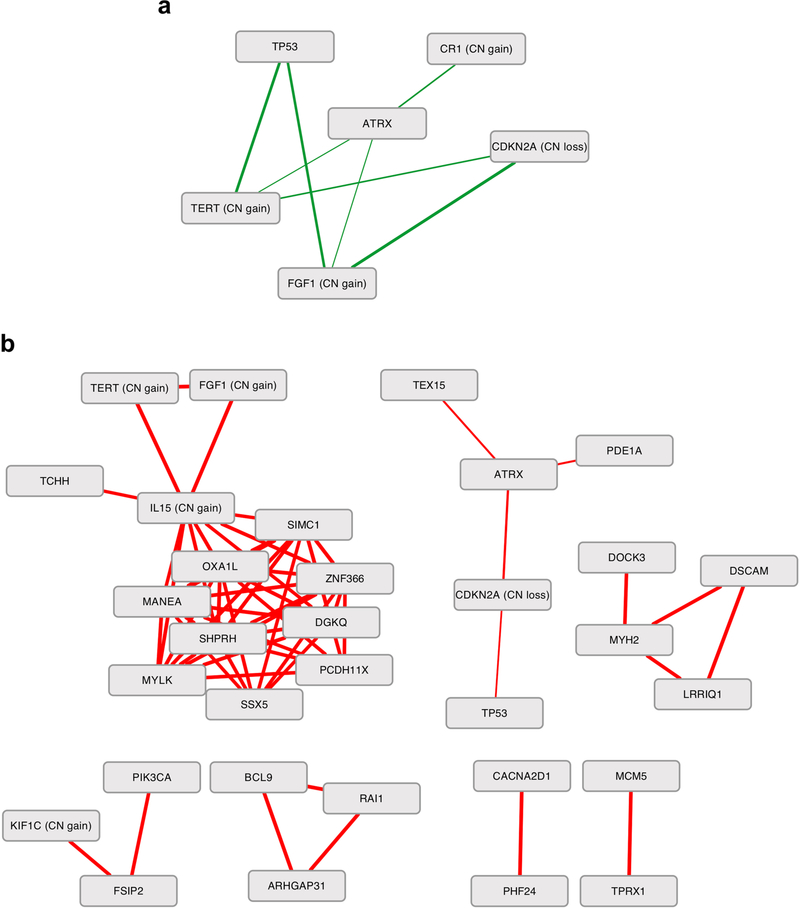
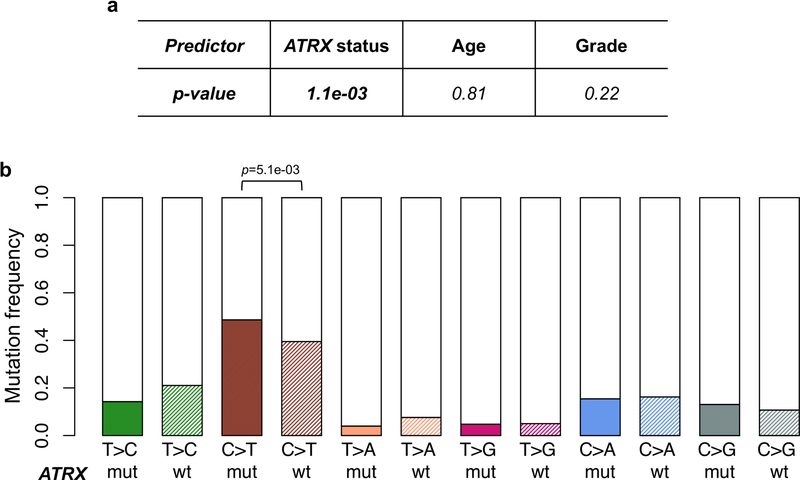
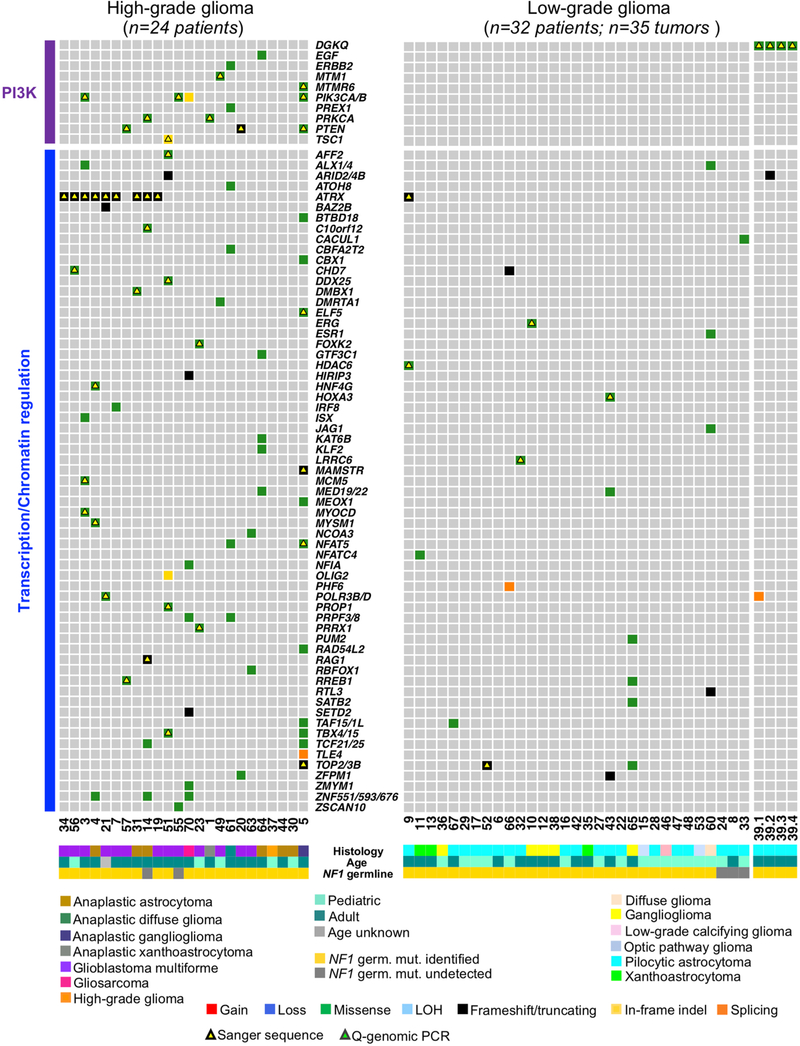
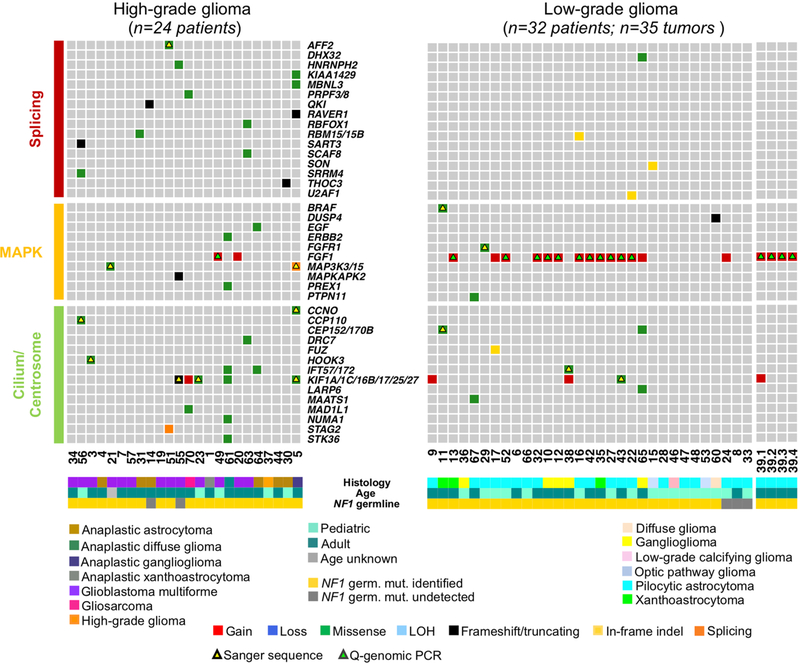
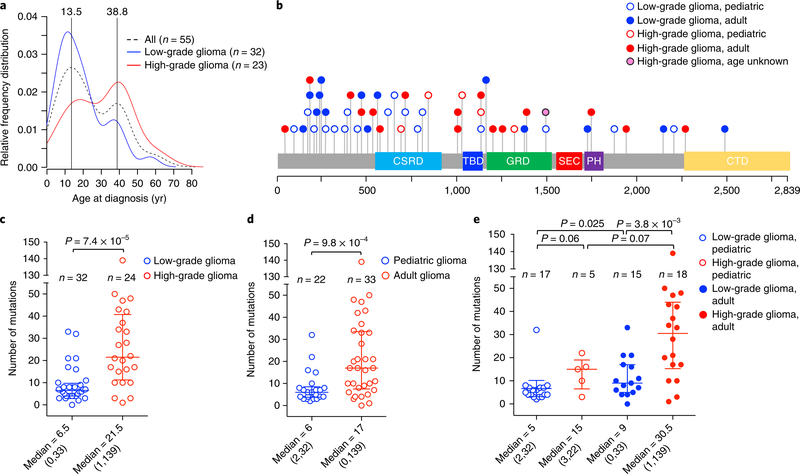
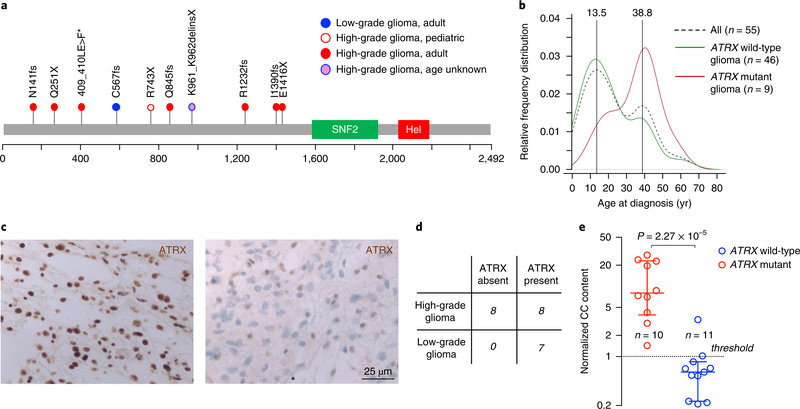
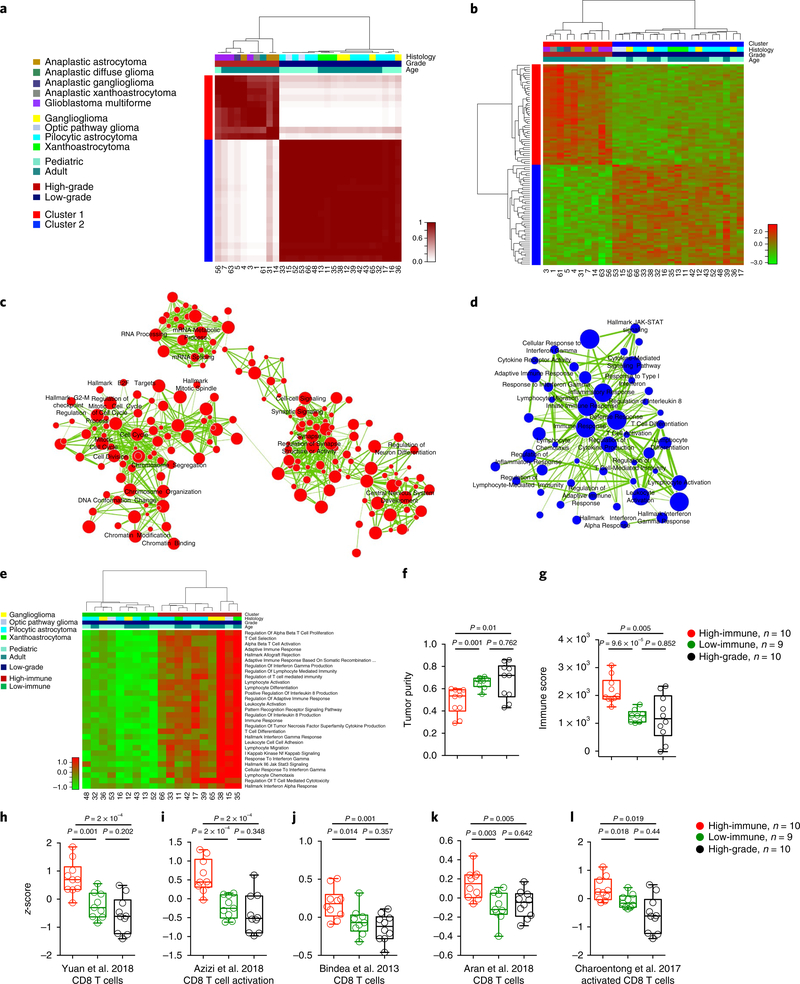
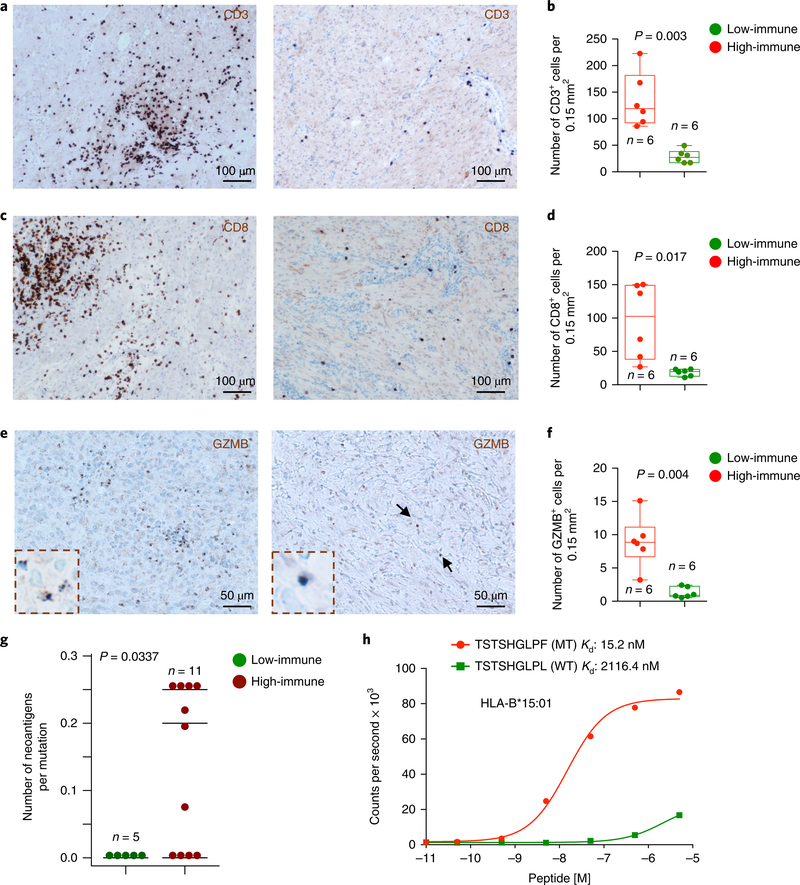
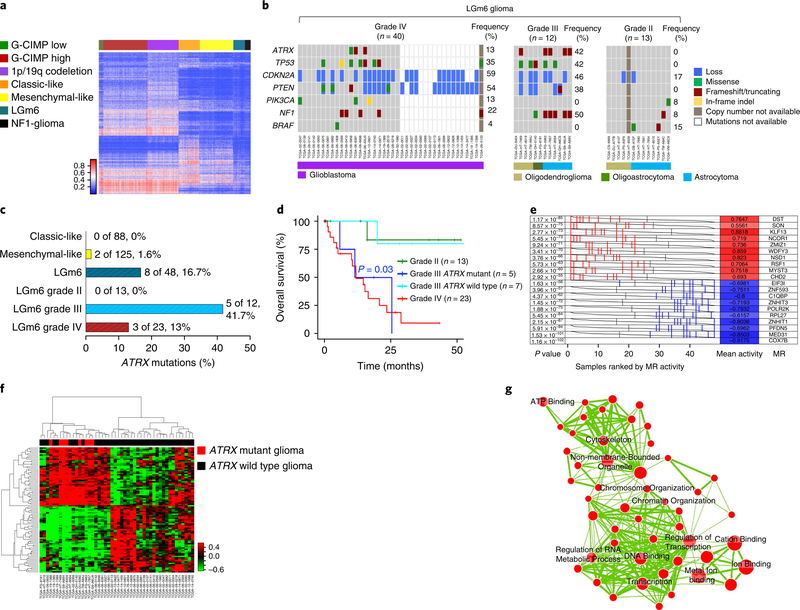
Neurofibromatosis type 1 (NF1) is a common tumor predisposition syndrome in which glioma is one of the prevalent tumors. Gliomagenesis in NF1 results in a heterogeneous spectrum of low- to high-grade neoplasms occurring during the entire lifespan of patients. The pattern of genetic and epigenetic alterations of glioma that develops in NF1 patients and the similarities with sporadic glioma remain unknown. Here, we present the molecular landscape of low- and high-grade gliomas in patients affected by NF1 (NF1-glioma). We found that the predisposing germline mutation of the NF1 gene was frequently converted to homozygosity and the somatic mutational load of NF1-glioma was influenced by age and grade. High-grade tumors harbored genetic alterations of TP53 and CDKN2A, frequent mutations of ATRX associated with Alternative Lengthening of Telomere, and were enriched in genetic alterations of transcription/chromatin regulation and PI3 kinase pathways. Low-grade tumors exhibited fewer mutations that were over-represented in genes of the MAP kinase pathway. Approximately 50% of low-grade NF1-gliomas displayed an immune signature, T lymphocyte infiltrates, and increased neo-antigen load. DNA methylation assigned NF1-glioma to LGm6, a poorly defined Isocitrate Dehydrogenase 1 wild-type subgroup enriched with ATRX mutations. Thus, the profiling of NF1-glioma defined a distinct landscape that recapitulates a subset of sporadic tumors.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures


References
-
- Uusitalo E. et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J. Invest. Dermatol 135, 904–906 (2015). - PubMed
-
- Evans DG et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am. J. Med. Genet. A 152A, 327–332 (2010). - PubMed
-
- Gutmann DH et al. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 3, 17004 (2017). - PubMed
-
- Brems H, Beert E, de Ravel T & Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 10, 508–515 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA127643/CA/NCI NIH HHS/United States
- R01 CA085628/CA/NCI NIH HHS/United States
- R01 CA190891/CA/NCI NIH HHS/United States
- R01 CA101644/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U54 CA193313/CA/NCI NIH HHS/United States
- R01 NS061776/NS/NINDS NIH HHS/United States
- R01 CA239698/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- R01 CA179044/CA/NCI NIH HHS/United States
- R01 CA131126/CA/NCI NIH HHS/United States
- R01 CA178546/CA/NCI NIH HHS/United States
- R01 CA239721/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
