

Identifying disease genes using machine learning and gene functional similarities, assessed through Gene Ontology
- PMID: 30532199
- PMCID: PMC6287949
- DOI: 10.1371/journal.pone.0208626
Identifying disease genes using machine learning and gene functional similarities, assessed through Gene Ontology
Abstract
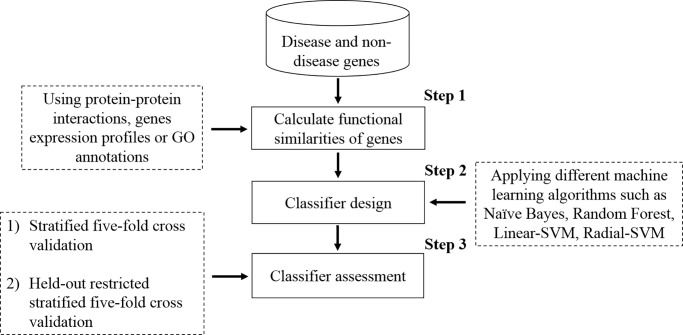
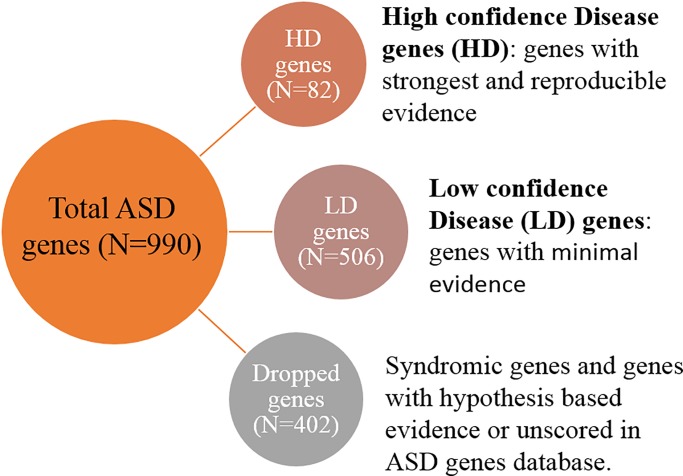
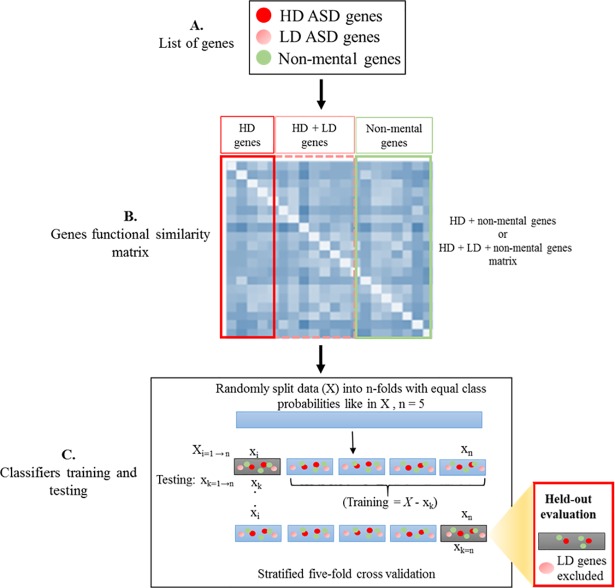
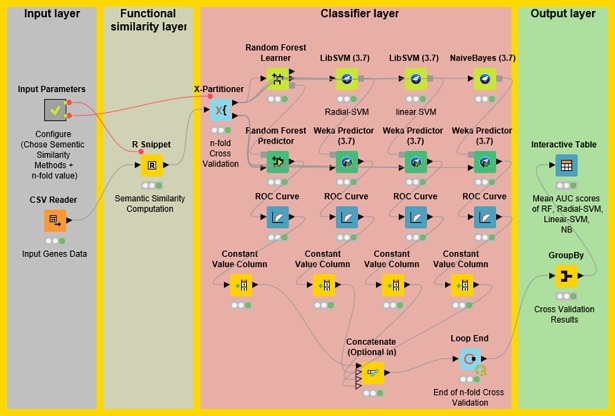
Identifying disease genes from a vast amount of genetic data is one of the most challenging tasks in the post-genomic era. Also, complex diseases present highly heterogeneous genotype, which difficult biological marker identification. Machine learning methods are widely used to identify these markers, but their performance is highly dependent upon the size and quality of available data. In this study, we demonstrated that machine learning classifiers trained on gene functional similarities, using Gene Ontology (GO), can improve the identification of genes involved in complex diseases. For this purpose, we developed a supervised machine learning methodology to predict complex disease genes. The proposed pipeline was assessed using Autism Spectrum Disorder (ASD) candidate genes. A quantitative measure of gene functional similarities was obtained by employing different semantic similarity measures. To infer the hidden functional similarities between ASD genes, various types of machine learning classifiers were built on quantitative semantic similarity matrices of ASD and non-ASD genes. The classifiers trained and tested on ASD and non-ASD gene functional similarities outperformed previously reported ASD classifiers. For example, a Random Forest (RF) classifier achieved an AUC of 0. 80 for predicting new ASD genes, which was higher than the reported classifier (0.73). Additionally, this classifier was able to predict 73 novel ASD candidate genes that were enriched for core ASD phenotypes, such as autism and obsessive-compulsive behavior. In addition, predicted genes were also enriched for ASD co-occurring conditions, including Attention Deficit Hyperactivity Disorder (ADHD). We also developed a KNIME workflow with the proposed methodology which allows users to configure and execute it without requiring machine learning and programming skills. Machine learning is an effective and reliable technique to decipher ASD mechanism by identifying novel disease genes, but this study further demonstrated that their performance can be improved by incorporating a quantitative measure of gene functional similarities. Source code and the workflow of the proposed methodology are available at https://github.com/Muh-Asif/ASD-genes-prediction.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Sanders SJ. First glimpses of the neurobiology of autism spectrum disorder. Curr Opin Genet Dev. Elsevier Ltd; 2015;33: 80–92. 10.1016/j.gde.2015.10.002 - DOI - PubMed
-
- Europe PMC Funders Group. Biological Insights From 108 Schizophrenia-Associated Genetic Loci. Nature. 2014;511: 421–427. 10.1038/nature13595 - DOI - PMC - PubMed
-
- Geschwind DH, State MW. Gene hunting in autism spectrum disorder: On the path to precision medicine. The Lancet Neurology. 2015. pp. 1109–1120. 10.1016/S1474-4422(15)00044-7 - DOI - PMC - PubMed
-
- Le DH, Kwon YK. GPEC: A Cytoscape plug-in for random walk-based gene prioritization and biomedical evidence collection. Comput Biol Chem. 2012;37: 17–23. 10.1016/j.compbiolchem.2012.02.004 - DOI - PubMed
-
- Peng J, Bai K, Shang X, Wang G, Xue H, Jin S, et al. Predicting disease-related genes using integrated biomedical networks. BMC Genomics. 2017;18 10.1186/s12864-016-3397-4 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
