

Predicting protein targets for drug-like compounds using transcriptomics
- PMID: 30532261
- PMCID: PMC6300300
- DOI: 10.1371/journal.pcbi.1006651
Predicting protein targets for drug-like compounds using transcriptomics
Abstract
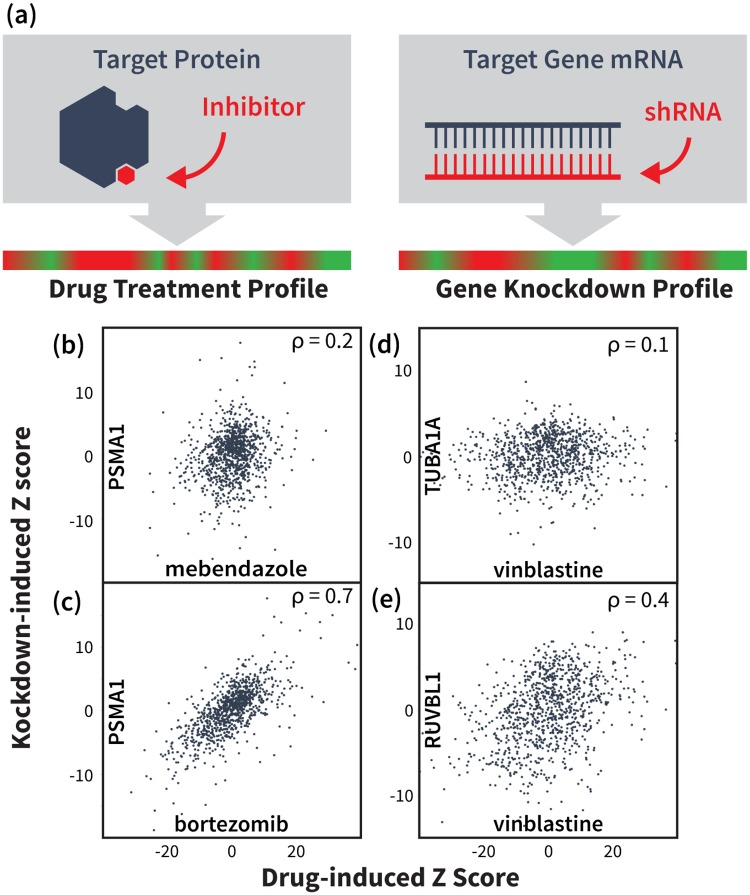
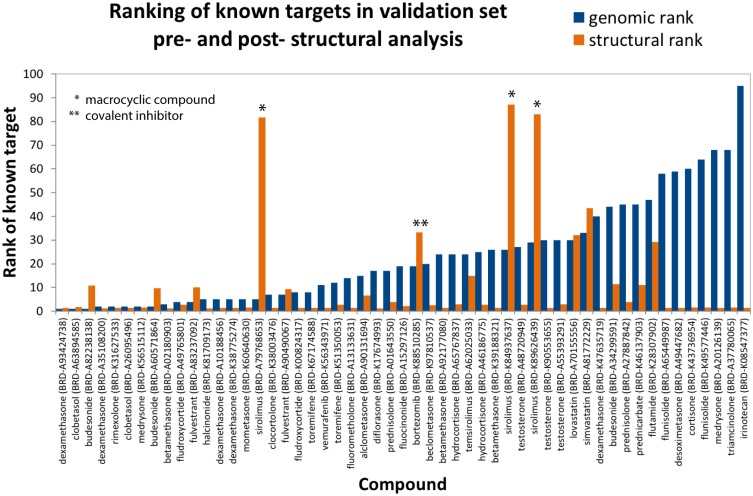
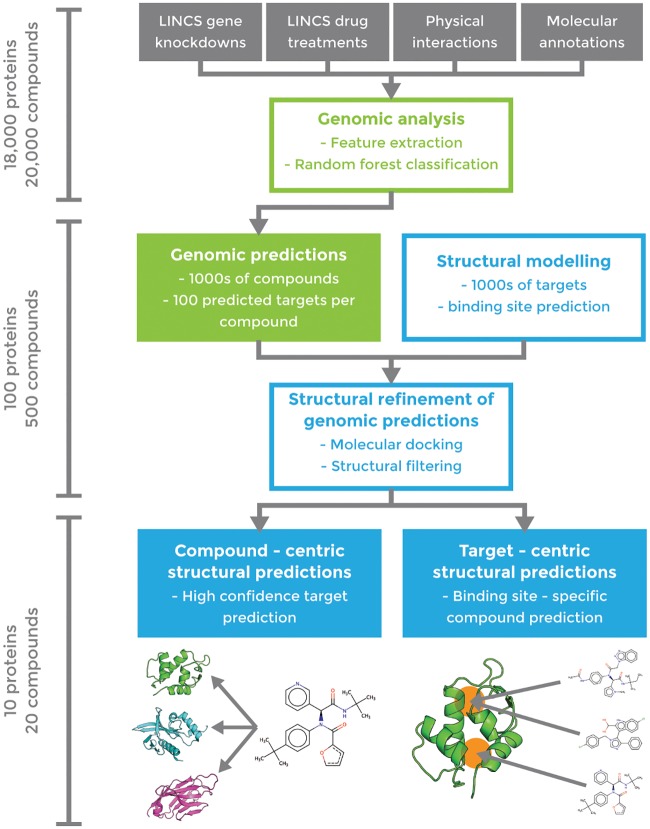
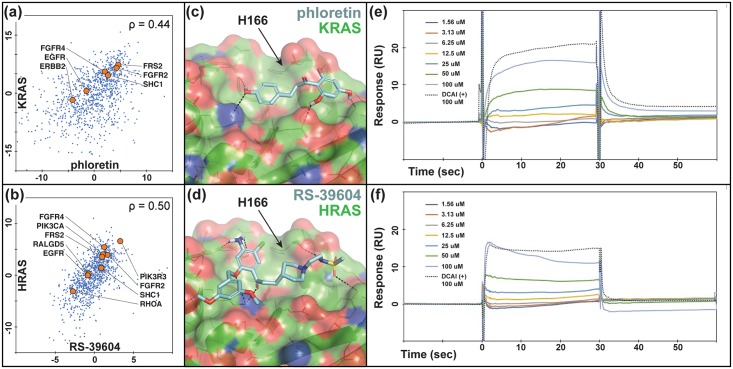
An expanded chemical space is essential for improved identification of small molecules for emerging therapeutic targets. However, the identification of targets for novel compounds is biased towards the synthesis of known scaffolds that bind familiar protein families, limiting the exploration of chemical space. To change this paradigm, we validated a new pipeline that identifies small molecule-protein interactions and works even for compounds lacking similarity to known drugs. Based on differential mRNA profiles in multiple cell types exposed to drugs and in which gene knockdowns (KD) were conducted, we showed that drugs induce gene regulatory networks that correlate with those produced after silencing protein-coding genes. Next, we applied supervised machine learning to exploit drug-KD signature correlations and enriched our predictions using an orthogonal structure-based screen. As a proof-of-principle for this regimen, top-10/top-100 target prediction accuracies of 26% and 41%, respectively, were achieved on a validation of set 152 FDA-approved drugs and 3104 potential targets. We then predicted targets for 1680 compounds and validated chemical interactors with four targets that have proven difficult to chemically modulate, including non-covalent inhibitors of HRAS and KRAS. Importantly, drug-target interactions manifest as gene expression correlations between drug treatment and both target gene KD and KD of genes that act up- or down-stream of the target, even for relatively weak binders. These correlations provide new insights on the cellular response of disrupting protein interactions and highlight the complex genetic phenotypes of drug treatment. With further refinement, our pipeline may accelerate the identification and development of novel chemical classes by screening compound-target interactions.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
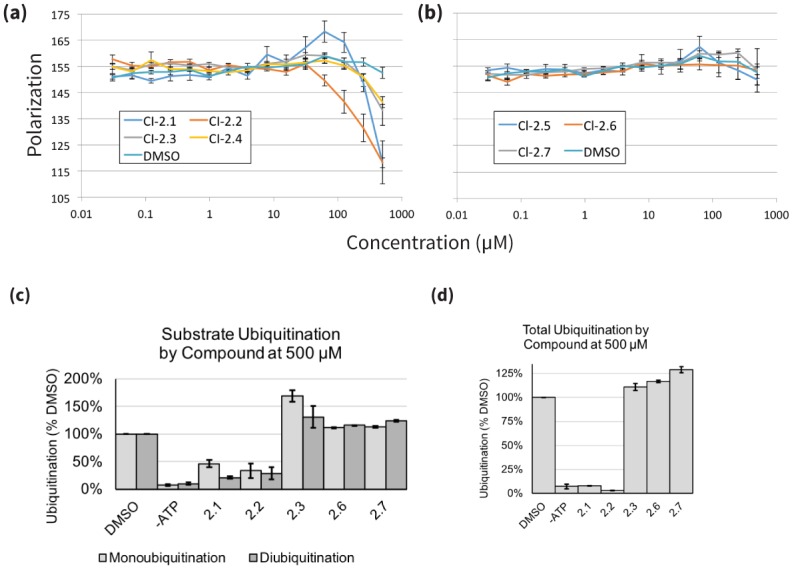
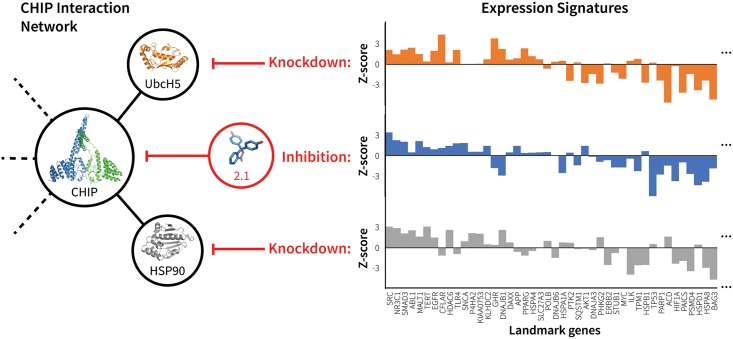
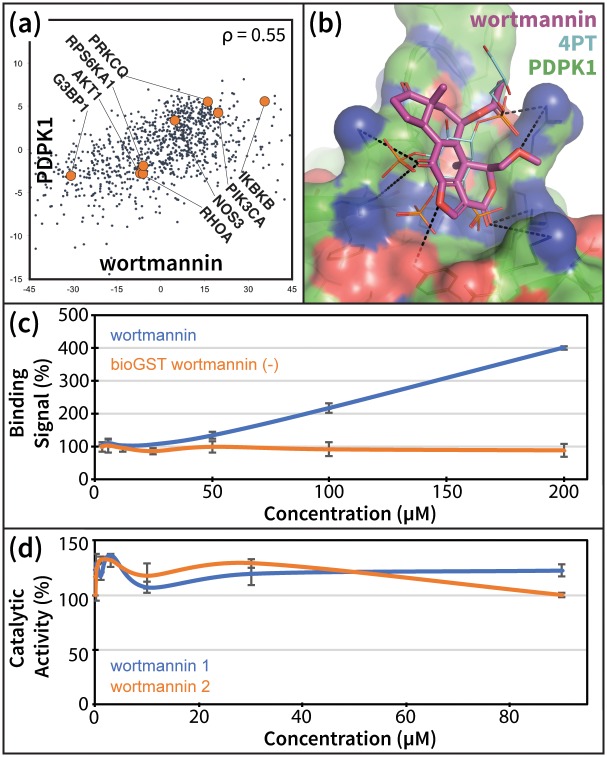
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
