

Iron in the Tumor Microenvironment-Connecting the Dots
- PMID: 30534534
- PMCID: PMC6275298
- DOI: 10.3389/fonc.2018.00549
Iron in the Tumor Microenvironment-Connecting the Dots
Abstract
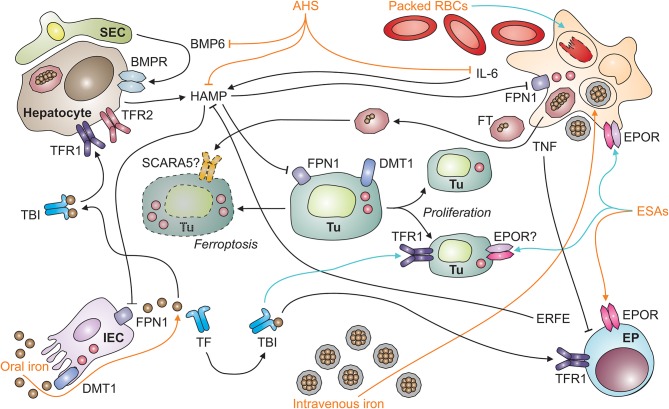
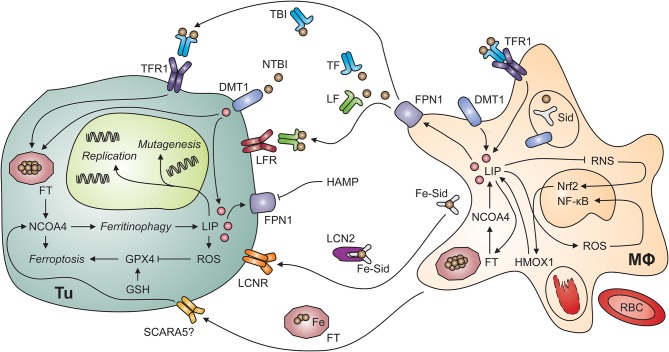
Iron metabolism and tumor biology are intimately linked. Iron facilitates the production of oxygen radicals, which may either result in iron-induced cell death, ferroptosis, or contribute to mutagenicity and malignant transformation. Once transformed, malignant cells require high amounts of iron for proliferation. In addition, iron has multiple regulatory effects on the immune system, thus affecting tumor surveillance by immune cells. For these reasons, inconsiderate iron supplementation in cancer patients has the potential of worsening disease course and outcome. On the other hand, chronic immune activation in the setting of malignancy alters systemic iron homeostasis and directs iron fluxes into myeloid cells. While this response aims at withdrawing iron from tumor cells, it may impair the effector functions of tumor-associated macrophages and will result in iron-restricted erythropoiesis and the development of anemia, subsequently. This review summarizes our current knowledge of the interconnections of iron homeostasis with cancer biology, discusses current clinical controversies in the treatment of anemia of cancer and focuses on the potential roles of iron in the solid tumor microenvironment, also speculating on yet unknown molecular mechanisms.
Keywords: ACD; TAM; anemia of cancer; ferroptosis; hepcidin; iron.
Figures
References
-
- Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. (1997) 272:19095–8. - PubMed
Publication types
LinkOut - more resources
Full Text Sources