

RELACS nuclei barcoding enables high-throughput ChIP-seq
- PMID: 30534606
- PMCID: PMC6281648
- DOI: 10.1038/s42003-018-0219-z
RELACS nuclei barcoding enables high-throughput ChIP-seq
Abstract
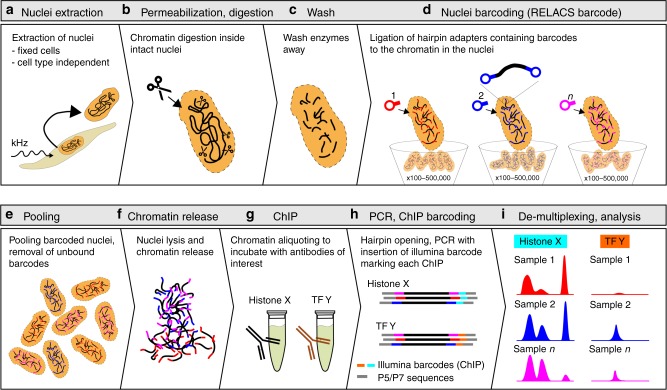
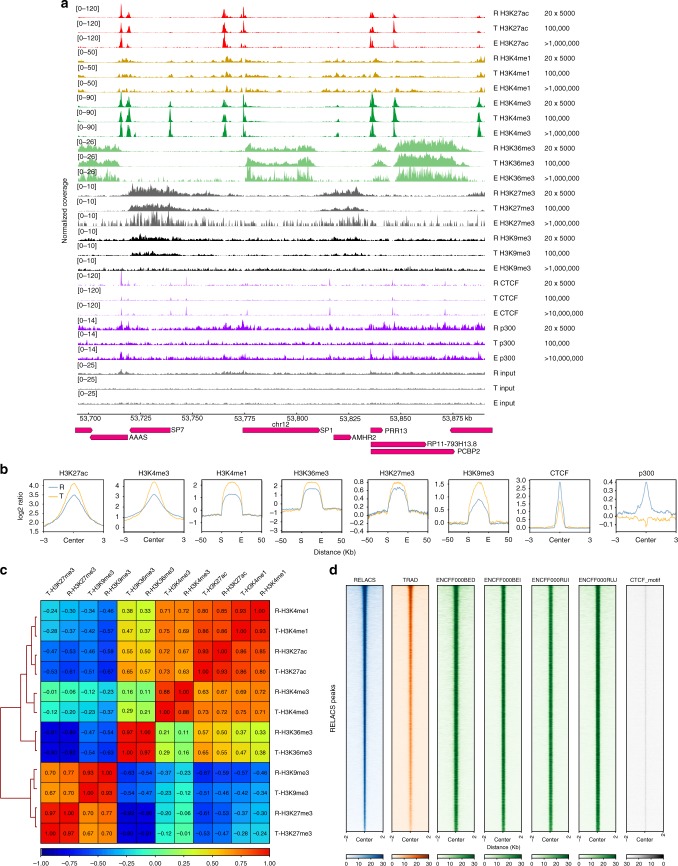
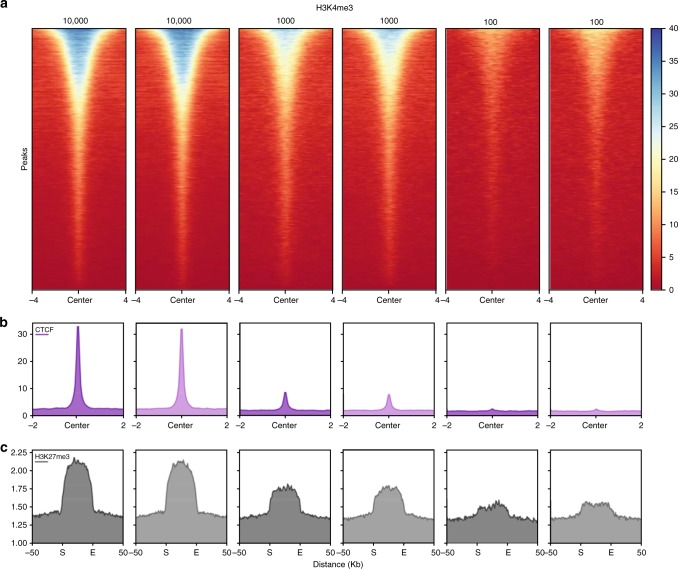
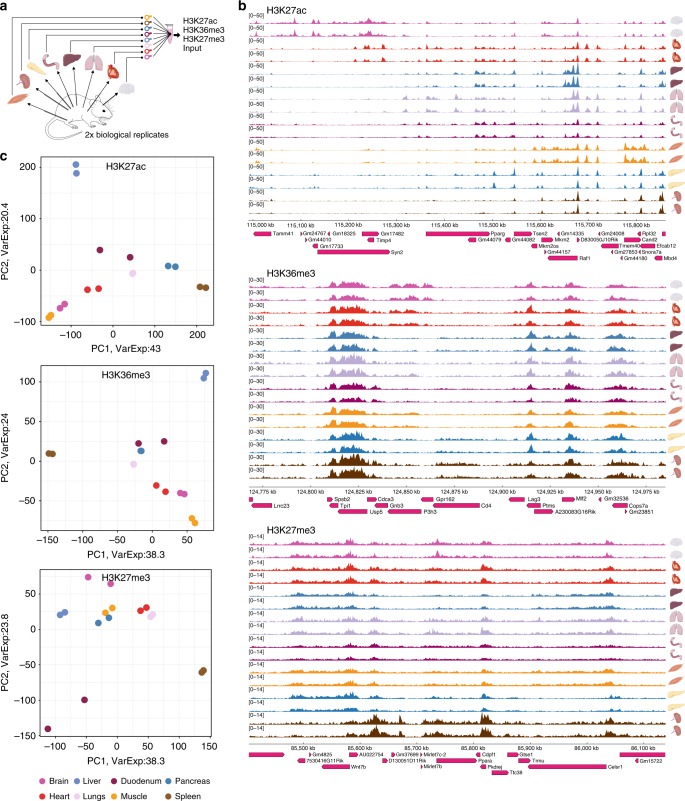
Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) is an invaluable tool for mapping chromatin-associated proteins. Current barcoding strategies aim to improve assay throughput and scalability but intense sample handling and lack of standardization over cell types, cell numbers and epitopes hinder wide-spread use in the field. Here, we present a barcoding method to enable high-throughput ChIP-seq using common molecular biology techniques. The method, called RELACS (restriction enzyme-based labeling of chromatin in situ) relies on standardized nuclei extraction from any source and employs chromatin cutting and barcoding within intact nuclei. Barcoded nuclei are pooled and processed within the same ChIP reaction, for maximal comparability and workload reduction. The innovative barcoding concept is particularly user-friendly and suitable for implementation to standardized large-scale clinical studies and scarce samples. Aiming to maximize universality and scalability, RELACS can generate ChIP-seq libraries for transcription factors and histone modifications from hundreds of samples within three days.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
