

Prot-SpaM: fast alignment-free phylogeny reconstruction based on whole-proteome sequences
- PMID: 30535314
- PMCID: PMC6436989
- DOI: 10.1093/gigascience/giy148
Prot-SpaM: fast alignment-free phylogeny reconstruction based on whole-proteome sequences
Abstract
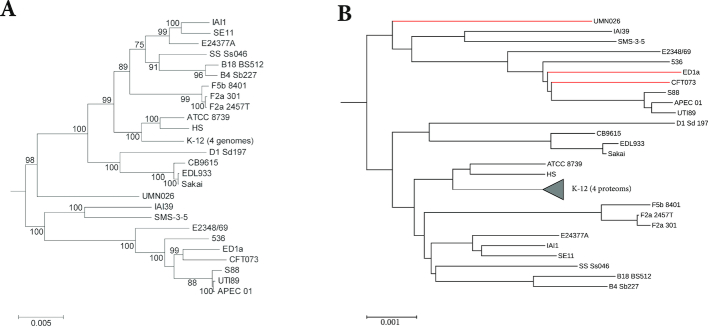
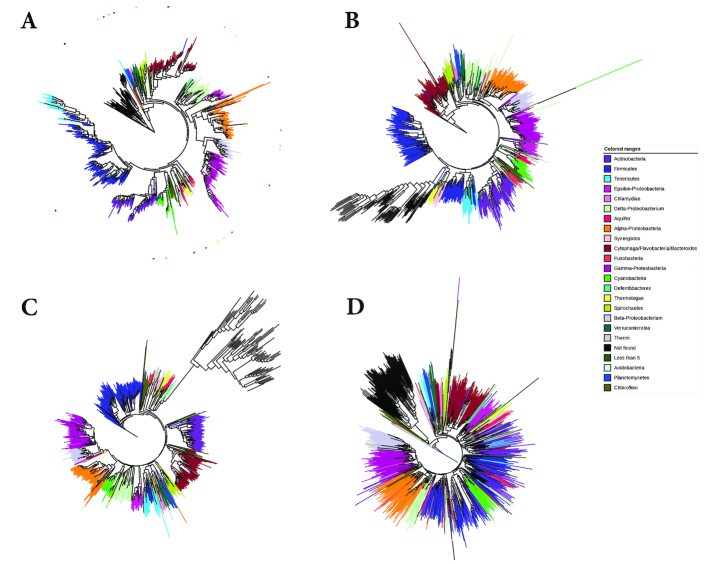
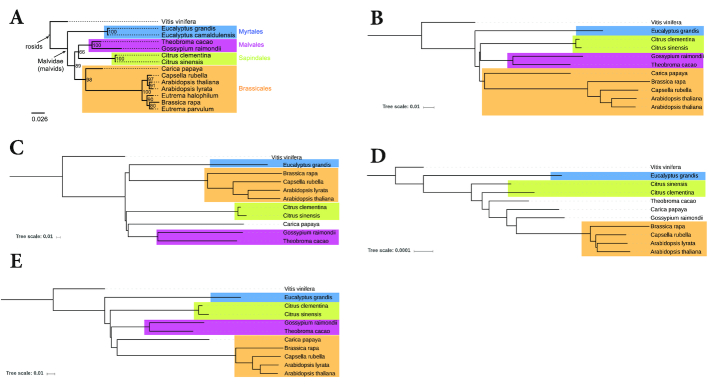
Word-based or 'alignment-free' sequence comparison has become an active research area in bioinformatics. While previous word-frequency approaches calculated rough measures of sequence similarity or dissimilarity, some new alignment-free methods are able to accurately estimate phylogenetic distances between genomic sequences. One of these approaches is Filtered Spaced Word Matches. Here, we extend this approach to estimate evolutionary distances between complete or incomplete proteomes; our implementation of this approach is called Prot-SpaM. We compare the performance of Prot-SpaM to other alignment-free methods on simulated sequences and on various groups of eukaryotic and prokaryotic taxa. Prot-SpaM can be used to calculate high-quality phylogenetic trees for dozens of whole-proteome sequences in a matter of seconds or minutes and often outperforms other alignment-free approaches. The source code of our software is available through Github: https://github.com/jschellh/ProtSpaM.
Keywords: Kimura; Wolbachia; alignment-free; amino-acid substitutions; distance method; micro-alignment; phylogeny; protein comparison; proteome; spaced words.
© The Author(s) 2018. Published by Oxford University Press.
Figures
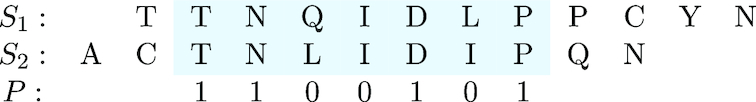
Similar articles
-
The number of k-mer matches between two DNA sequences as a function of k and applications to estimate phylogenetic distances.PLoS One. 2020 Feb 10;15(2):e0228070. doi: 10.1371/journal.pone.0228070. eCollection 2020. PLoS One. 2020. PMID: 32040534 Free PMC article.
-
Fast and accurate phylogeny reconstruction using filtered spaced-word matches.Bioinformatics. 2017 Apr 1;33(7):971-979. doi: 10.1093/bioinformatics/btw776. Bioinformatics. 2017. PMID: 28073754 Free PMC article.
-
Sequence Comparison Without Alignment: The SpaM Approaches.Methods Mol Biol. 2021;2231:121-134. doi: 10.1007/978-1-0716-1036-7_8. Methods Mol Biol. 2021. PMID: 33289890
-
Read-SpaM: assembly-free and alignment-free comparison of bacterial genomes with low sequencing coverage.BMC Bioinformatics. 2019 Dec 17;20(Suppl 20):638. doi: 10.1186/s12859-019-3205-7. BMC Bioinformatics. 2019. PMID: 31842735 Free PMC article.
-
Alignment-free phylogenetics and population genetics.Brief Bioinform. 2014 May;15(3):407-18. doi: 10.1093/bib/bbt083. Epub 2013 Nov 29. Brief Bioinform. 2014. PMID: 24291823 Review.
Cited by
-
AlcoR: alignment-free simulation, mapping, and visualization of low-complexity regions in biological data.Gigascience. 2022 Dec 28;12:giad101. doi: 10.1093/gigascience/giad101. Epub 2023 Dec 13. Gigascience. 2022. PMID: 38091509 Free PMC article.
-
SWeeP: representing large biological sequences datasets in compact vectors.Sci Rep. 2020 Jan 9;10(1):91. doi: 10.1038/s41598-019-55627-4. Sci Rep. 2020. PMID: 31919449 Free PMC article.
-
Evolutionary Insight into the Trypanosomatidae Using Alignment-Free Phylogenomics of the Kinetoplast.Pathogens. 2019 Sep 18;8(3):157. doi: 10.3390/pathogens8030157. Pathogens. 2019. PMID: 31540520 Free PMC article.
-
The number of k-mer matches between two DNA sequences as a function of k and applications to estimate phylogenetic distances.PLoS One. 2020 Feb 10;15(2):e0228070. doi: 10.1371/journal.pone.0228070. eCollection 2020. PLoS One. 2020. PMID: 32040534 Free PMC article.
-
CGRWDL: alignment-free phylogeny reconstruction method for viruses based on chaos game representation weighted by dynamical language model.Front Microbiol. 2024 Mar 20;15:1339156. doi: 10.3389/fmicb.2024.1339156. eCollection 2024. Front Microbiol. 2024. PMID: 38572227 Free PMC article.
References
-
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–74. - PubMed
-
- Liu L, Xi Z, Wu S, et al. .. Estimating phylogenetic trees from genome-scale data. Annals of the New York Academy of Sciences. 2015;1360:36–53. - PubMed
-
- Bininda-Emonds ORP. The evolution of supertrees. Trends in Ecology and Evolution. 2004;19:315–22. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
