

Mutant p53 as a guardian of the cancer cell
- PMID: 30538286
- PMCID: PMC6329812
- DOI: 10.1038/s41418-018-0246-9
Mutant p53 as a guardian of the cancer cell
Abstract
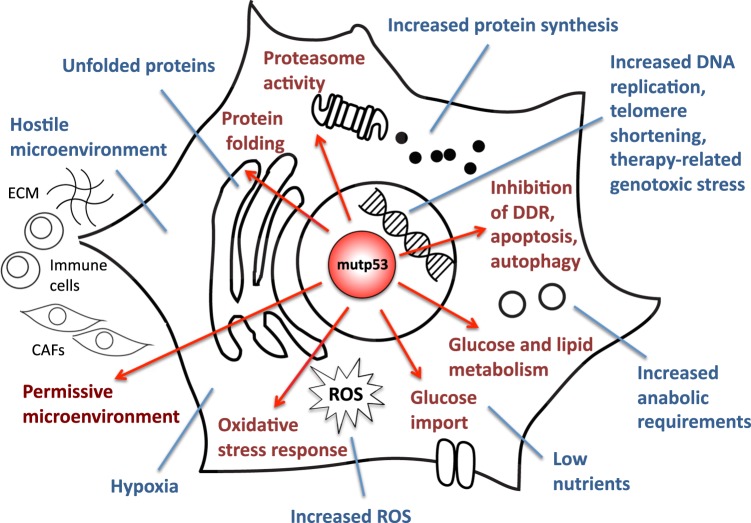
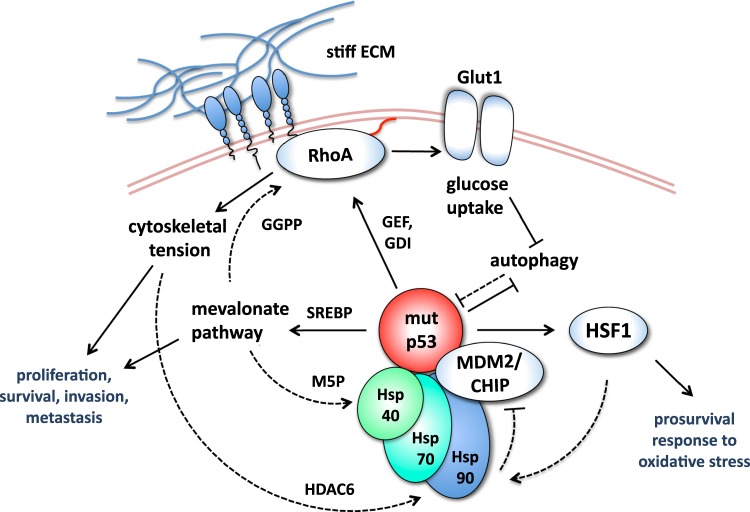
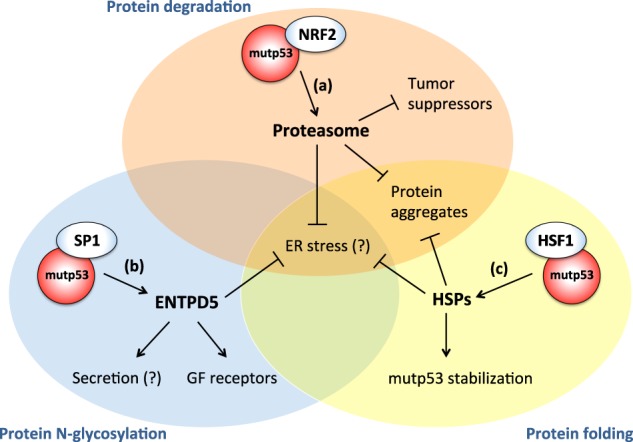
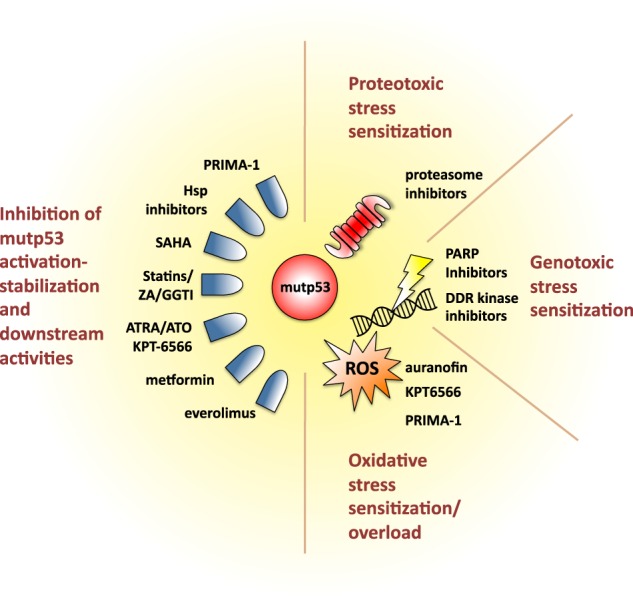
Forty years of research have established that the p53 tumor suppressor provides a major barrier to neoplastic transformation and tumor progression by its unique ability to act as an extremely sensitive collector of stress inputs, and to coordinate a complex framework of diverse effector pathways and processes that protect cellular homeostasis and genome stability. Missense mutations in the TP53 gene are extremely widespread in human cancers and give rise to mutant p53 proteins that lose tumor suppressive activities, and some of which exert trans-dominant repression over the wild-type counterpart. Cancer cells acquire selective advantages by retaining mutant forms of the protein, which radically subvert the nature of the p53 pathway by promoting invasion, metastasis and chemoresistance. In this review, we consider available evidence suggesting that mutant p53 proteins can favor cancer cell survival and tumor progression by acting as homeostatic factors that sense and protect cancer cells from transformation-related stress stimuli, including DNA lesions, oxidative and proteotoxic stress, metabolic inbalance, interaction with the tumor microenvironment, and the immune system. These activities of mutant p53 may explain cancer cell addiction to this particular oncogene, and their study may disclose tumor vulnerabilities and synthetic lethalities that could be exploited for hitting tumors bearing missense TP53 mutations.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
