

Factor cooperation for chromosome discrimination in Drosophila
- PMID: 30541149
- PMCID: PMC6393291
- DOI: 10.1093/nar/gky1238
Factor cooperation for chromosome discrimination in Drosophila
Abstract
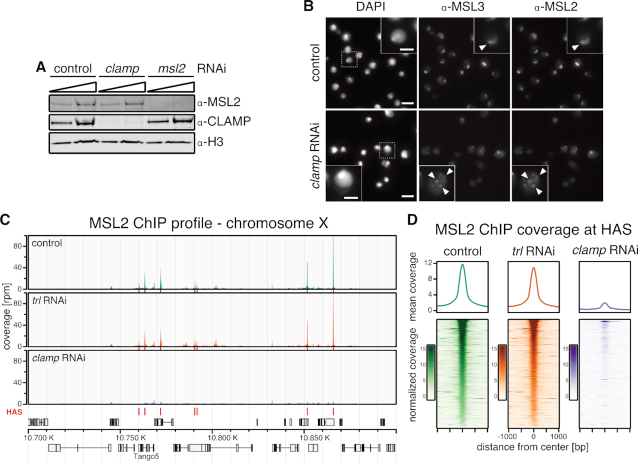
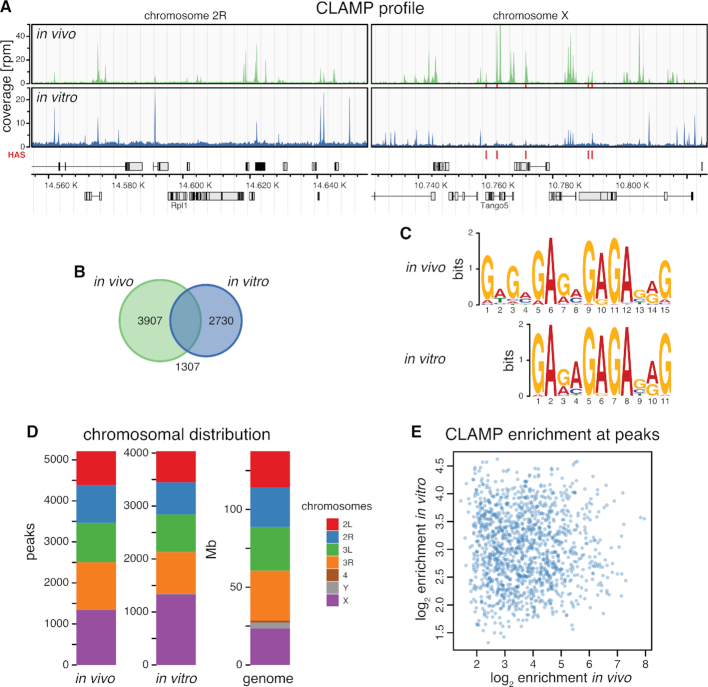
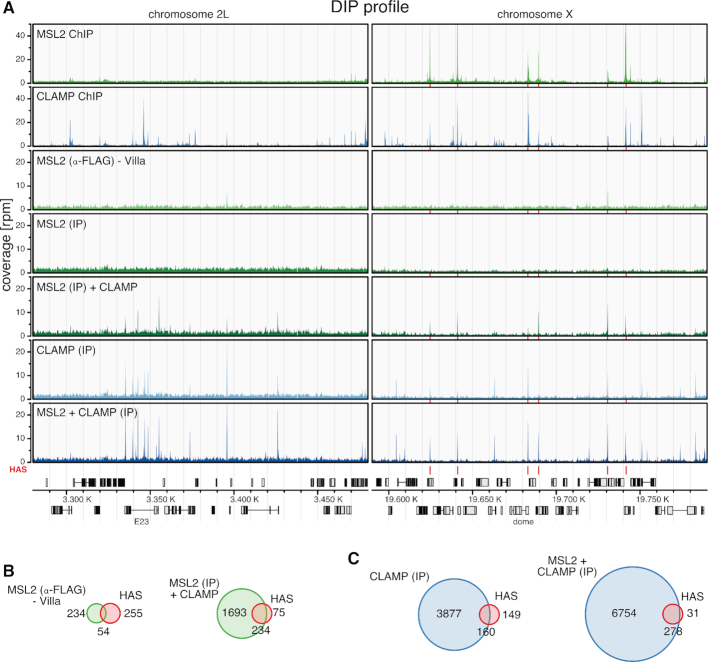
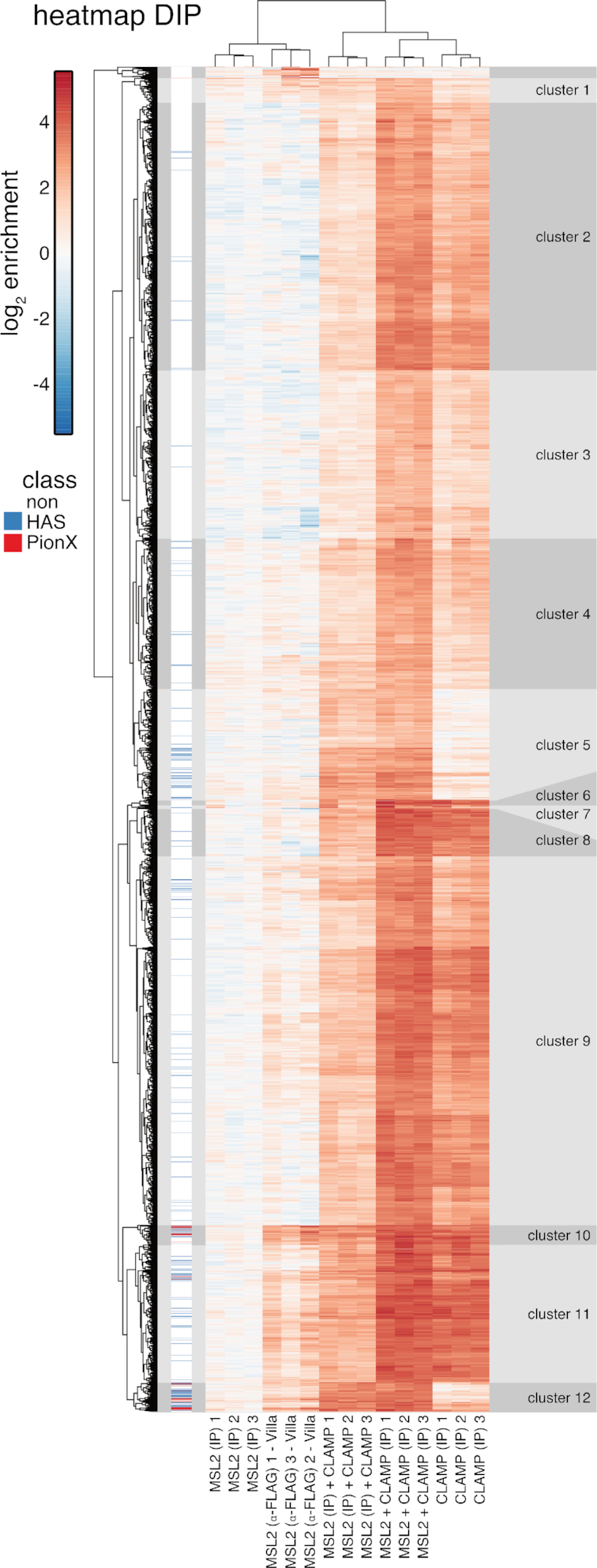
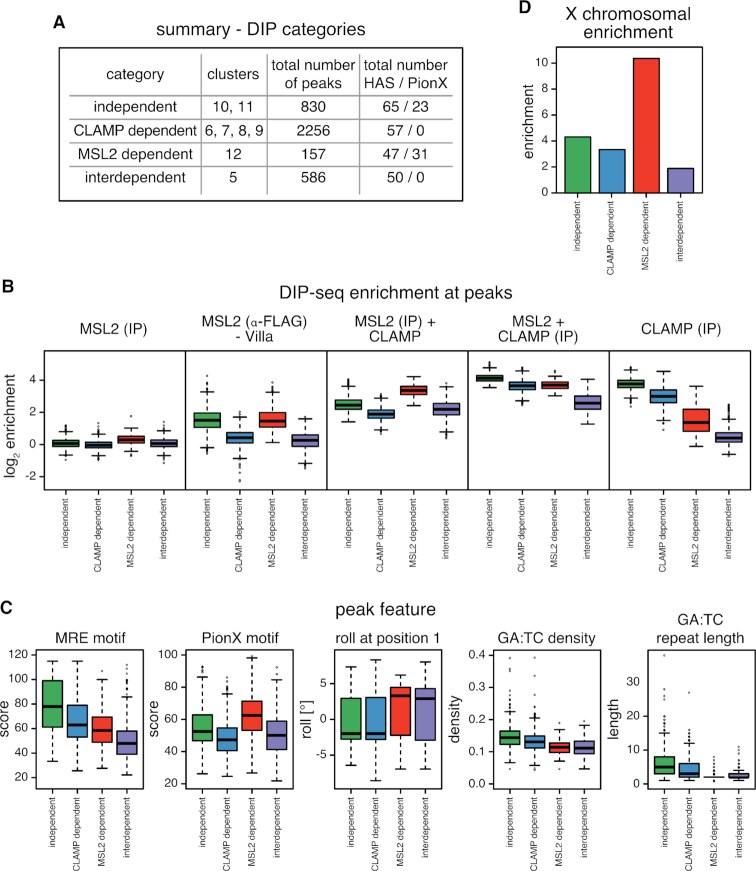
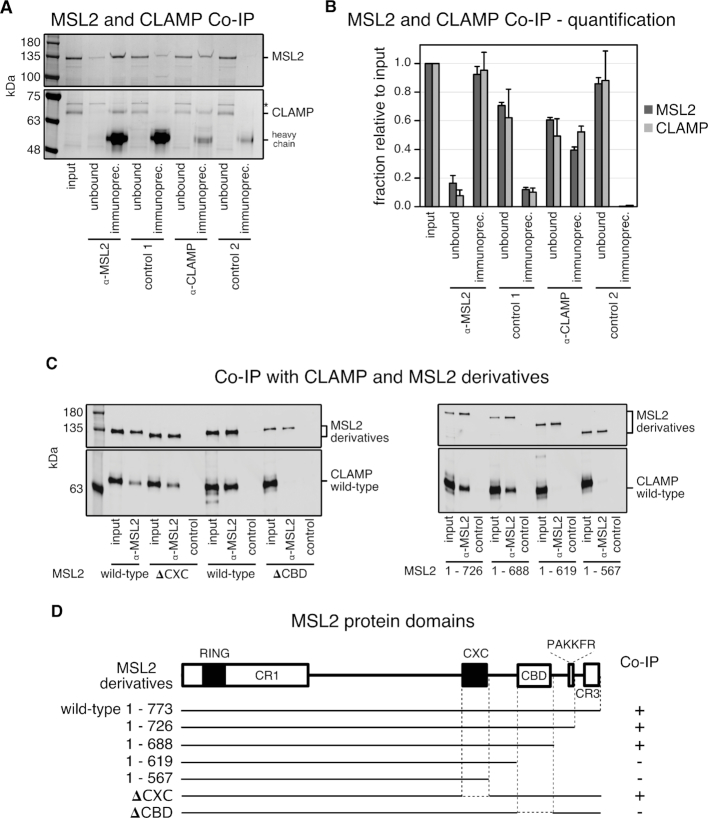
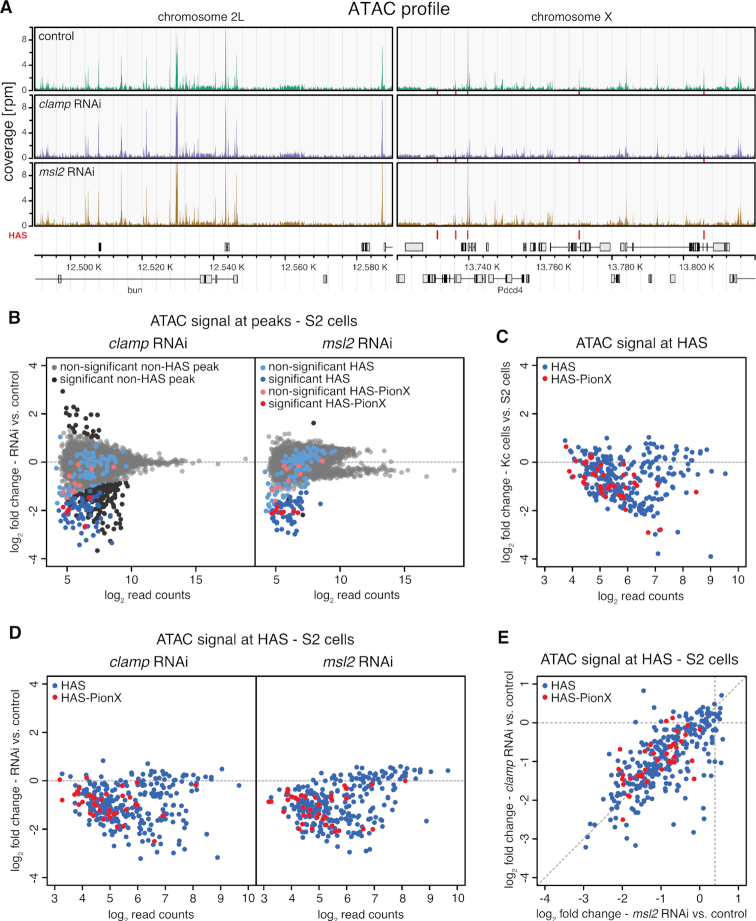
Transcription regulators select their genomic binding sites from a large pool of similar, non-functional sequences. Although general principles that allow such discrimination are known, the complexity of DNA elements often precludes a prediction of functional sites. The process of dosage compensation in Drosophila allows exploring the rules underlying binding site selectivity. The male-specific-lethal (MSL) Dosage Compensation Complex (DCC) selectively binds to some 300 X chromosomal 'High Affinity Sites' (HAS) containing GA-rich 'MSL recognition elements' (MREs), but disregards thousands of other MRE sequences in the genome. The DNA-binding subunit MSL2 alone identifies a subset of MREs, but fails to recognize most MREs within HAS. The 'Chromatin-linked adaptor for MSL proteins' (CLAMP) also interacts with many MREs genome-wide and promotes DCC binding to HAS. Using genome-wide DNA-immunoprecipitation we describe extensive cooperativity between both factors, depending on the nature of the binding sites. These are explained by physical interaction between MSL2 and CLAMP. In vivo, both factors cooperate to compete with nucleosome formation at HAS. The male-specific MSL2 thus synergises with a ubiquitous GA-repeat binding protein for refined X/autosome discrimination.
© The Author(s) 2018. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Jolma A., Yin Y., Nitta K.R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J.. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015; 527:384–388. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
