

Development of Stable Rotavirus Reporter Expression Systems
- PMID: 30541830
- PMCID: PMC6363997
- DOI: 10.1128/JVI.01774-18
Development of Stable Rotavirus Reporter Expression Systems
Abstract
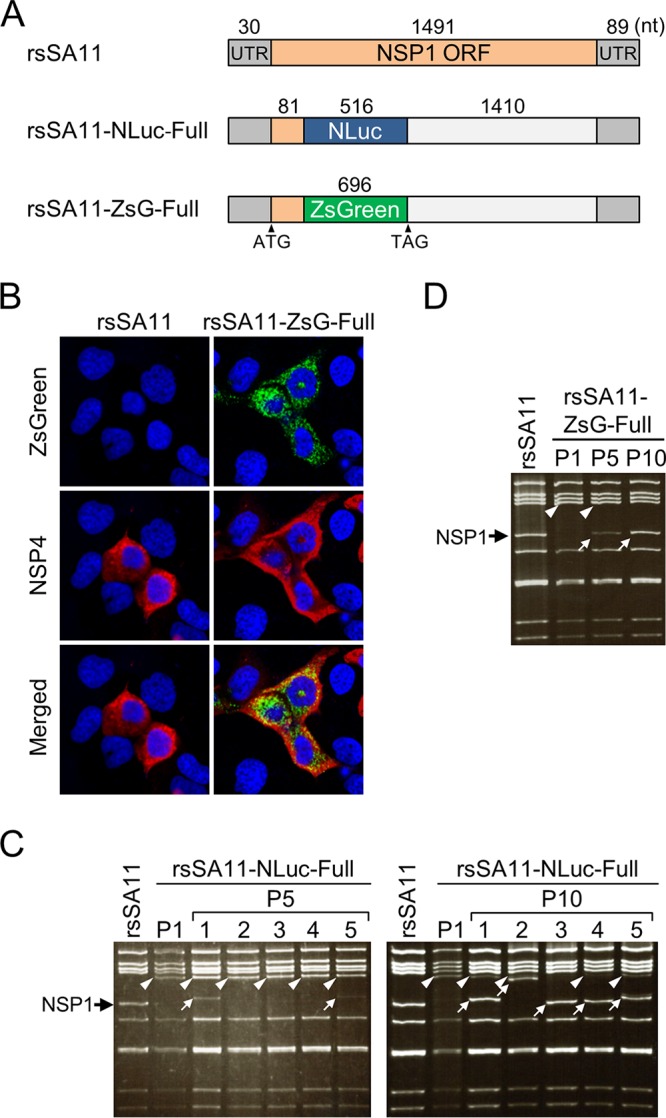
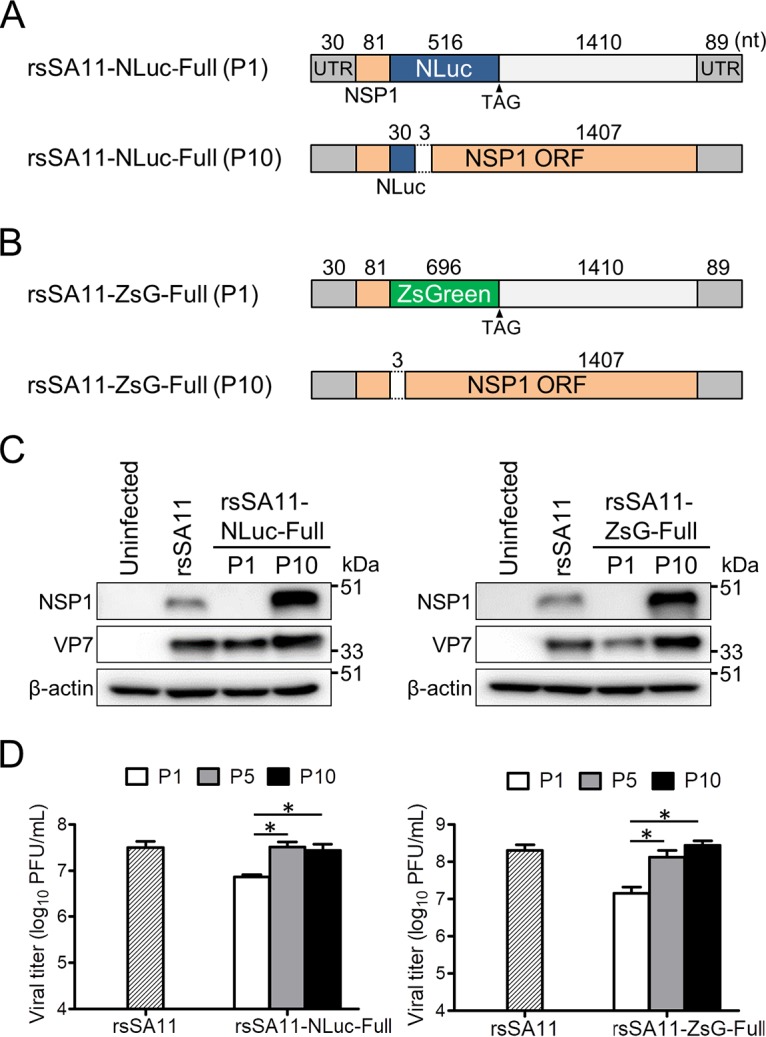
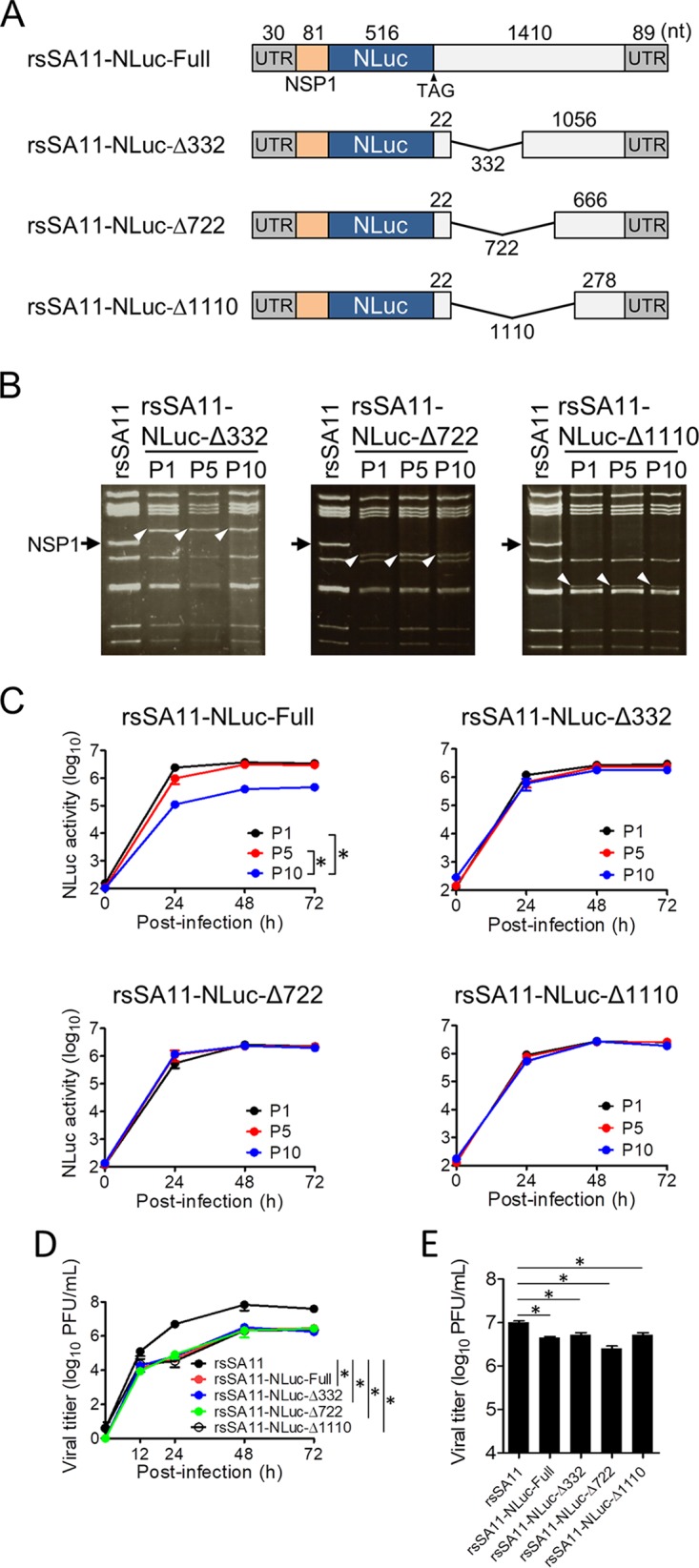
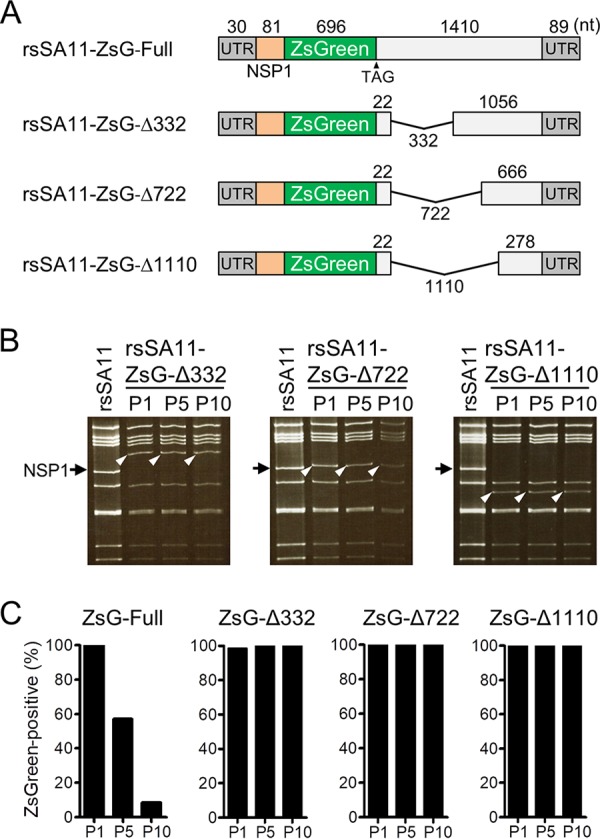
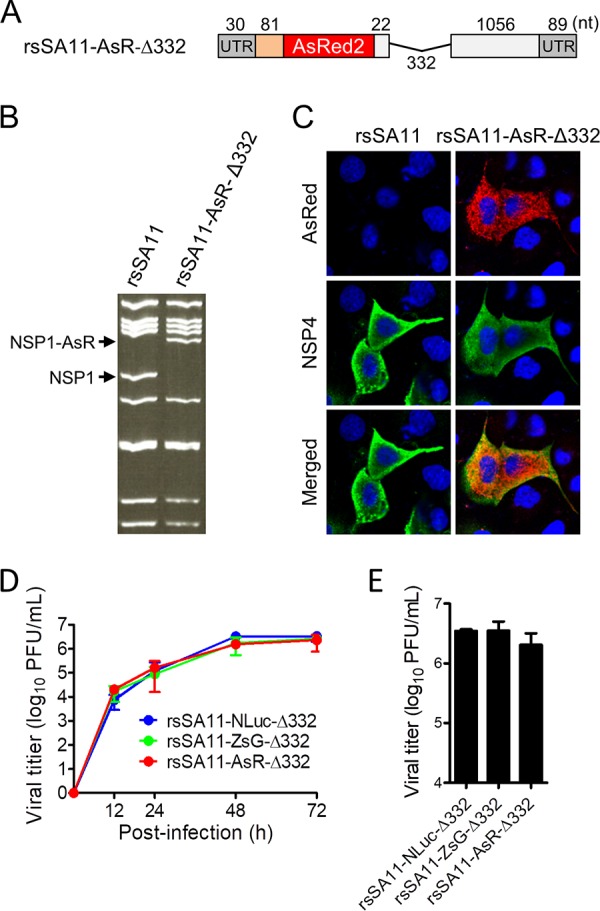
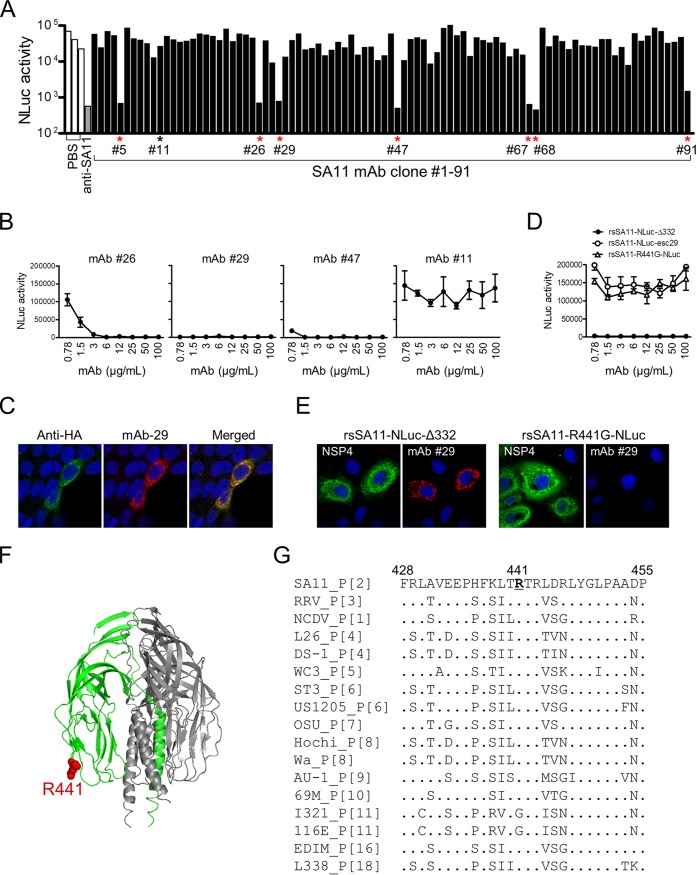
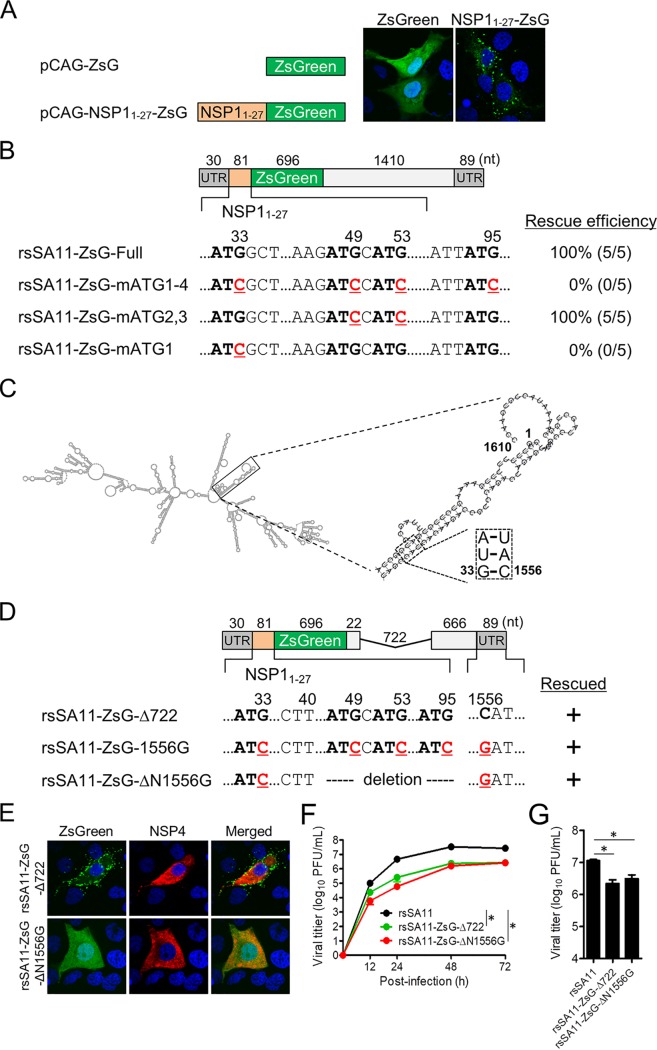
Engineered recombinant viruses expressing reporter genes have been developed for real-time monitoring of replication and for mass screening of antiviral inhibitors. Recently, we reported using a reverse genetics system to develop the first recombinant reporter rotaviruses (RVs) that expressed NanoLuc (NLuc) luciferase. Here, we describe a strategy for developing stable reporter RVs expressing luciferase and green or red fluorescent proteins. The reporter genes were inserted into the open reading frame of NSP1 and expressed as a fusion with an NSP1 peptide consisting of amino acids 1 to 27. The stability of foreign genes within the reporter RV strains harboring a shorter chimeric NSP1-reporter gene was greater than that of those in the original reporter RV strain, independent of the transgene inserted. The improved reporter RV was used to screen for neutralizing monoclonal antibodies (MAbs). Sequence analysis of escape mutants from one MAb clone (clone 29) identified an amino acid substitution (arginine to glycine) at position 441 in the VP4 protein, which resides within neutralizing epitope 5-1 in the VP5* fragment. Furthermore, to express a native reporter protein lacking NSP1 amino acids 1 to 27, the 5'- and 3'-terminal region sequences were modified to restore the predicted secondary RNA structure of the NSP1-reporter chimeric gene. These data demonstrate the utility of reporter RVs for live monitoring of RV infections and also suggest further applications (e.g., RV vaccine vectors, which can induce mucosal immunity against intestinal pathogens).IMPORTANCE Development of reporter RVs has been hampered by the lack of comprehensive reverse genetics systems. Recently, we developed a plasmid-based reverse genetics system that enables generation of reporter RVs expressing NLuc luciferase. The prototype reporter RV had some disadvantages (i.e., the transgene was unstable and was expressed as a fusion protein with a partial NSP1 peptide); however, the improved reporter RV overcomes these problems through modification of the untranslated region of the reporter-NSP1 chimeric gene. This strategy for generating stable reporter RVs could be expanded to diverse transgenes and be used to develop RV transduction vectors. Also, the data improve our understanding of the importance of 5'- and 3'-terminal sequences in terms of genome replication, assembly, and packaging.
Keywords: reporter virus; rotavirus; virus vector.
Copyright © 2019 American Society for Microbiology.
Figures
Similar articles
-
Generation of recombinant rotaviruses encoding a split NanoLuc peptide tag.Biochem Biophys Res Commun. 2021 Jan 1;534:740-746. doi: 10.1016/j.bbrc.2020.11.007. Epub 2020 Nov 26. Biochem Biophys Res Commun. 2021. PMID: 33250174
-
Generation of Recombinant Rotavirus Expressing NSP3-UnaG Fusion Protein by a Simplified Reverse Genetics System.J Virol. 2019 Nov 26;93(24):e01616-19. doi: 10.1128/JVI.01616-19. Print 2019 Dec 15. J Virol. 2019. PMID: 31597761 Free PMC article.
-
Reverse Genetics System for a Human Group A Rotavirus.J Virol. 2020 Jan 6;94(2):e00963-19. doi: 10.1128/JVI.00963-19. Print 2020 Jan 6. J Virol. 2020. PMID: 31645445 Free PMC article.
-
Rotaviruses and rotavirus vaccines.Br Med Bull. 2009;90:37-51. doi: 10.1093/bmb/ldp009. Epub 2009 Feb 20. Br Med Bull. 2009. PMID: 19233929 Review.
-
Rotavirus reverse genetics: A tool for understanding virus biology.Virus Res. 2021 Nov;305:198576. doi: 10.1016/j.virusres.2021.198576. Epub 2021 Sep 21. Virus Res. 2021. PMID: 34560180 Review.
Cited by
-
U-CAN-seq: A Universal Competition Assay by Nanopore Sequencing.Viruses. 2024 Apr 19;16(4):636. doi: 10.3390/v16040636. Viruses. 2024. PMID: 38675976 Free PMC article.
-
Generation of single-round infectious rotavirus with a mutation in the intermediate capsid protein VP6.J Virol. 2024 Jul 23;98(7):e0076224. doi: 10.1128/jvi.00762-24. Epub 2024 Jun 5. J Virol. 2024. PMID: 38837379 Free PMC article.
-
Development of an entirely plasmid-based reverse genetics system for 12-segmented double-stranded RNA viruses.Proc Natl Acad Sci U S A. 2021 Oct 19;118(42):e2105334118. doi: 10.1073/pnas.2105334118. Proc Natl Acad Sci U S A. 2021. PMID: 34635593 Free PMC article.
-
Characterization of Sialic Acid-Independent Simian Rotavirus Mutants in Viral Infection and Pathogenesis.J Virol. 2023 Jan 31;97(1):e0139722. doi: 10.1128/jvi.01397-22. Epub 2023 Jan 5. J Virol. 2023. PMID: 36602365 Free PMC article.
-
Generation of Simian Rotavirus Reassortants with VP4- and VP7-Encoding Genome Segments from Human Strains Circulating in Africa Using Reverse Genetics.Viruses. 2020 Feb 11;12(2):201. doi: 10.3390/v12020201. Viruses. 2020. PMID: 32054092 Free PMC article.
References
-
- McGee CE, Shustov AV, Tsetsarkin K, Frolov IV, Mason PW, Vanlandingham DL, Higgs S. 2010. Infection, dissemination, and transmission of a West Nile virus green fluorescent protein infectious clone by Culex pipiens quinquefasciatus mosquitoes. Vector Borne Zoonotic Dis 10:267–274. doi:10.1089/vbz.2009.0067. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
