

Insulin-Like Growth Factor 2 Receptor Expression Is Promoted by Human Herpesvirus 8-Encoded Interleukin-6 and Contributes to Viral Latency and Productive Replication
- PMID: 30541844
- PMCID: PMC6384062
- DOI: 10.1128/JVI.02026-18
Insulin-Like Growth Factor 2 Receptor Expression Is Promoted by Human Herpesvirus 8-Encoded Interleukin-6 and Contributes to Viral Latency and Productive Replication
Abstract
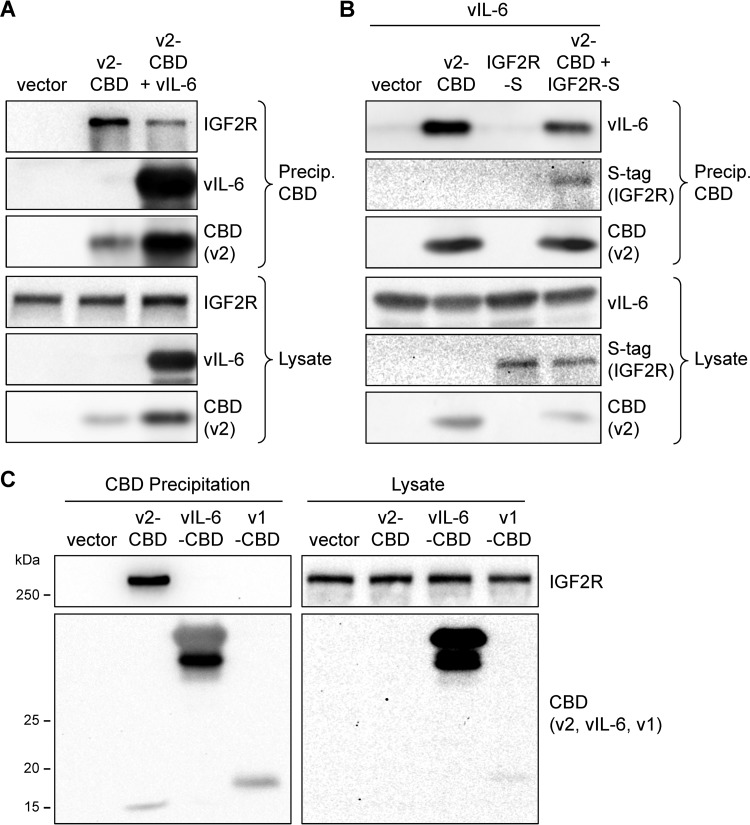
Human herpesvirus 8 (HHV-8) viral interleukin-6 (vIL-6) localizes largely to the endoplasmic reticulum (ER) and here associates functionally with both the gp130 signal transducer and the novel ER membrane protein vitamin K epoxide reductase complex subunit 1 variant-2 (VKORC1v2). The latter interaction contributes to the viability of latently infected primary effusion lymphoma (PEL) cells and to HHV-8 productive replication, in part via promotion of ER-associated degradation (ERAD) of nascent pro-cathepsin D (pCatD) and consequent suppression of lysosome-localized proapoptotic mature CatD. Here we report that VKORC1v2 associates with insulin-like growth factor 2 receptor (IGF2R), also known as cation-independent mannose-6-phosphate receptor, which is involved in trafficking of mannose-6-phosphate-conjugated glycoproteins to lysosomes. VKORC1v2 effected reduced IGF2R expression in a manner dependent on VKORC1v2-IGF2R interaction, while vIL-6, which could inhibit VKORC1v2-IGF2R interaction, effected increased expression of IGF2R. These effects were independent of changes in IGF2R mRNA levels, indicating likely posttranslational mechanisms. In kinetic analyses involving labeling of either newly synthesized or preexisting IGF2R, vIL-6 promoted accumulation of the former while having no detectable effect on the latter. Furthermore, vIL-6 led to decreased K48-linked ubiquitination of IGF2R and suppression of ERAD proteins effected increased IGF2R expression and loss of IGF2R regulation by vIL-6. Depletion-based experiments identified IGF2R as a promoter of PEL cell viability and virus yields from lytically reactivated cultures. Our findings identify ER-transiting nascent IGF2R as an interaction partner of VKORC1v2 and target of vIL-6 regulation and IGF2R as a positive contributor to HHV-8 biology, thereby extending understanding of the mechanisms of VKORC1v2-associated vIL-6 function.IMPORTANCE HHV-8 vIL-6 promotes productive replication in the context of reactivated lytic replication in primary effusion lymphoma (PEL) and endothelial cells and sustains latently infected PEL cell viability. Viral IL-6 is also considered to contribute significantly to HHV-8-associated pathogenesis, since vIL-6 can promote cell proliferation, cell survival, and angiogenesis that are characteristic of HHV-8-associated Kaposi's sarcoma, PEL and multicentric Castleman's disease (MCD), in addition to proinflammatory activities observed in MCD-like "Kaposi's sarcoma-associated herpesvirus-induced cytokine syndrome." We show in the present study that vIL-6 can promote productive replication and latent PEL cell viability through upregulation of the mannose-6-phosphate- and peptide hormone-interacting receptor IGF2R, which is a positive factor in HHV-8 biology via these activities. VKORC1v2-enhanced ER-associated degradation of IGF2R and vIL-6 promotion of IGF2R expression through prevention of its interaction with VKORC1v2 and consequent rescue from degradation represent newly recognized activities of VKOCR1v2 and vIL-6.
Keywords: ER-associated degradation; cation-independent mannose-6-phosphate receptor; endoplasmic reticulum; human herpesvirus 8; insulin-like growth factor 2 receptor; latency; replication; viral interleukin-6; vitamin K epoxide reductase complex subunit 1 variant-2.
Copyright © 2019 American Society for Microbiology.
Figures







Similar articles
-
Human Herpesvirus 8 Interleukin-6 Interacts with Calnexin Cycle Components and Promotes Protein Folding.J Virol. 2017 Oct 27;91(22):e00965-17. doi: 10.1128/JVI.00965-17. Print 2017 Nov 15. J Virol. 2017. PMID: 28878084 Free PMC article.
-
Promotion of Endoplasmic Reticulum-Associated Degradation of Procathepsin D by Human Herpesvirus 8-Encoded Viral Interleukin-6.J Virol. 2015 Aug;89(15):7979-90. doi: 10.1128/JVI.00375-15. Epub 2015 May 27. J Virol. 2015. PMID: 26018151 Free PMC article.
-
Human herpesvirus 8 interleukin-6 contributes to primary effusion lymphoma cell viability via suppression of proapoptotic cathepsin D, a cointeraction partner of vitamin K epoxide reductase complex subunit 1 variant 2.J Virol. 2014 Jan;88(2):1025-38. doi: 10.1128/JVI.02830-13. Epub 2013 Nov 6. J Virol. 2014. PMID: 24198402 Free PMC article.
-
Viral interleukin-6: role in Kaposi's sarcoma-associated herpesvirus: associated malignancies.J Interferon Cytokine Res. 2011 Nov;31(11):791-801. doi: 10.1089/jir.2011.0043. Epub 2011 Jul 18. J Interferon Cytokine Res. 2011. PMID: 21767154 Free PMC article. Review.
-
Viral Interleukin-6: Structure, pathophysiology and strategies of neutralization.Eur J Cell Biol. 2011 Jun-Jul;90(6-7):495-504. doi: 10.1016/j.ejcb.2010.10.016. Epub 2010 Dec 21. Eur J Cell Biol. 2011. PMID: 21176991 Review.
Cited by
-
Genetic Analyses of Contributions of Viral Interleukin-6 Interactions and Signaling to Human Herpesvirus 8 Productive Replication.J Virol. 2020 Sep 15;94(19):e00909-20. doi: 10.1128/JVI.00909-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32669340 Free PMC article.
-
Broadening Horizons: Exploring the Cathepsin Family as Therapeutic Targets for Alzheimer's Disease.Aging Dis. 2024 Jun 2;16(4):1987-2007. doi: 10.14336/AD.2024.0456. Aging Dis. 2024. PMID: 39122455 Free PMC article. Review.
-
Molecular Mechanisms of Kaposi Sarcoma-Associated Herpesvirus (HHV8)-Related Lymphomagenesis.Cancers (Basel). 2024 Oct 31;16(21):3693. doi: 10.3390/cancers16213693. Cancers (Basel). 2024. PMID: 39518131 Free PMC article. Review.
-
Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases.Microorganisms. 2020 Mar 11;8(3):388. doi: 10.3390/microorganisms8030388. Microorganisms. 2020. PMID: 32168836 Free PMC article. Review.
-
Polypharmacology-based kinome screen identifies new regulators of KSHV reactivation.PLoS Pathog. 2023 Sep 5;19(9):e1011169. doi: 10.1371/journal.ppat.1011169. eCollection 2023 Sep. PLoS Pathog. 2023. PMID: 37669313 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
