

SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy
- PMID: 30541864
- PMCID: PMC6340340
- DOI: 10.1212/WNL.0000000000006729
SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy
Erratum in
-
SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy.Neurology. 2019 Nov 12;93(20):908. doi: 10.1212/WNL.0000000000008352. Neurology. 2019. PMID: 31712380 Free PMC article. No abstract available.
Abstract
Objective: To delineate the epileptology, a key part of the SYNGAP1 phenotypic spectrum, in a large patient cohort.
Methods: Patients were recruited via investigators' practices or social media. We included patients with (likely) pathogenic SYNGAP1 variants or chromosome 6p21.32 microdeletions incorporating SYNGAP1. We analyzed patients' phenotypes using a standardized epilepsy questionnaire, medical records, EEG, MRI, and seizure videos.
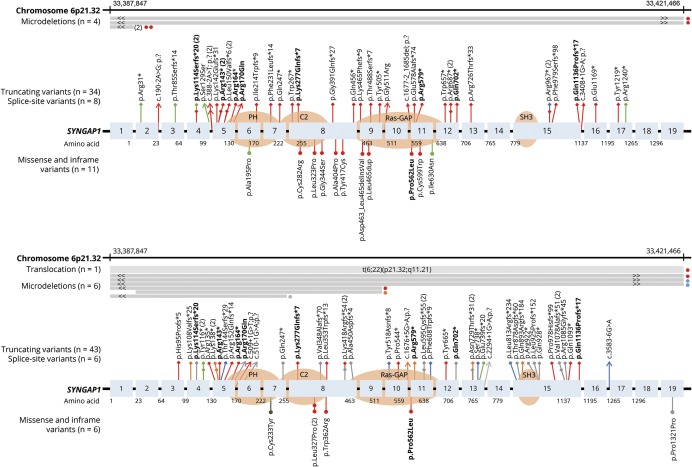
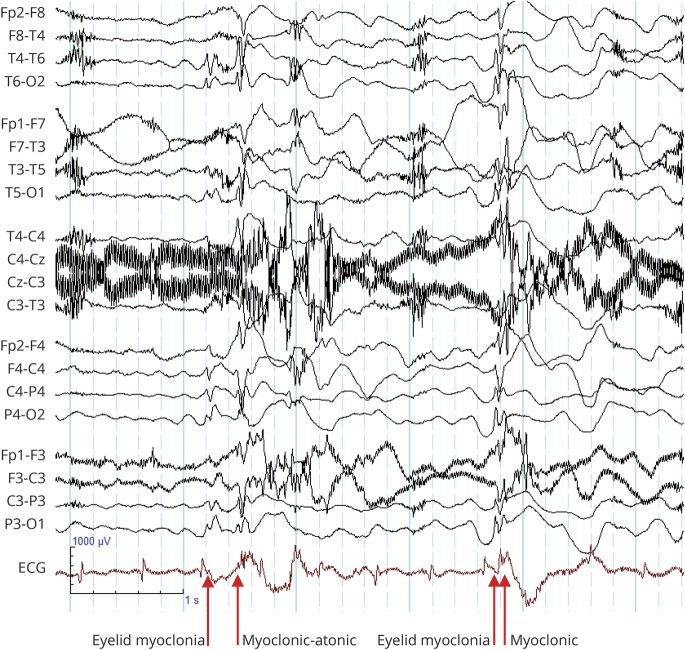
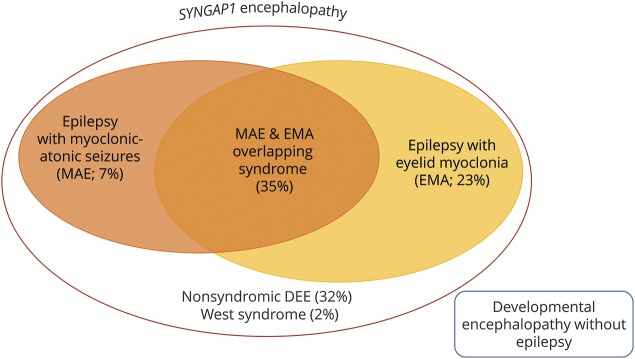
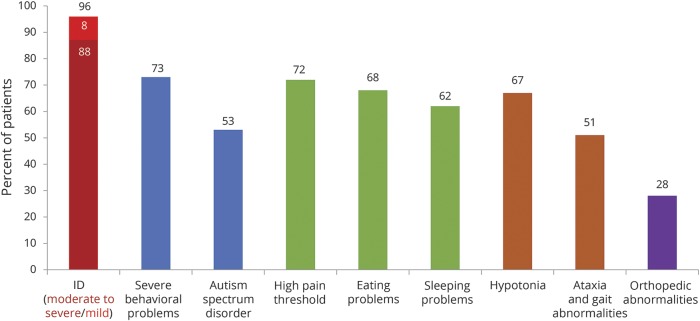
Results: We included 57 patients (53% male, median age 8 years) with SYNGAP1 mutations (n = 53) or microdeletions (n = 4). Of the 57 patients, 56 had epilepsy: generalized in 55, with focal seizures in 7 and infantile spasms in 1. Median seizure onset age was 2 years. A novel type of drop attack was identified comprising eyelid myoclonia evolving to a myoclonic-atonic (n = 5) or atonic (n = 8) seizure. Seizure types included eyelid myoclonia with absences (65%), myoclonic seizures (34%), atypical (20%) and typical (18%) absences, and atonic seizures (14%), triggered by eating in 25%. Developmental delay preceded seizure onset in 54 of 56 (96%) patients for whom early developmental history was available. Developmental plateauing or regression occurred with seizures in 56 in the context of a developmental and epileptic encephalopathy (DEE). Fifty-five of 57 patients had intellectual disability, which was moderate to severe in 50. Other common features included behavioral problems (73%); high pain threshold (72%); eating problems, including oral aversion (68%); hypotonia (67%); sleeping problems (62%); autism spectrum disorder (54%); and ataxia or gait abnormalities (51%).
Conclusions: SYNGAP1 mutations cause a generalized DEE with a distinctive syndrome combining epilepsy with eyelid myoclonia with absences and myoclonic-atonic seizures, as well as a predilection to seizures triggered by eating.
Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures

Comment in
-
A synaptic protein defect associated with reflex seizure disorder.Neurology. 2019 Jan 8;92(2):63-64. doi: 10.1212/WNL.0000000000006720. Epub 2018 Dec 12. Neurology. 2019. PMID: 30541867 No abstract available.
-
Reader response: SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy.Neurology. 2020 Feb 25;94(8):368-369. doi: 10.1212/WNL.0000000000009007. Neurology. 2020. PMID: 32094282 No abstract available.
-
Reader response: SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy.Neurology. 2020 Feb 25;94(8):369. doi: 10.1212/WNL.0000000000009009. Neurology. 2020. PMID: 32094283 No abstract available.
-
Author response: SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy.Neurology. 2020 Feb 25;94(8):370. doi: 10.1212/WNL.0000000000009010. Neurology. 2020. PMID: 32094284 No abstract available.
References
-
- Klitten LL, Møller RS, Nikanorova M, Silahtaroglu A, Hjalgrim H, Tommerup N. A balanced translocation disrupts SYNGAP1 in a patient with intellectual disability, speech impairment, and epilepsy with myoclonic absences (EMA). Epilepsia 2011;52:190–193. - PubMed
-
- Krepischi ACV, Rosenberg C, Costa SS, Crolla JA, Huang S, Vianna-Morgante AM. A novel de novo microdeletion spanning the SYNGAP1 gene on the short arm of chromosome 6 associated with mental retardation. Am J Med Genet A 2010;152A:2365–2378. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical