

Trapping biosynthetic acyl-enzyme intermediates with encoded 2,3-diaminopropionic acid
- PMID: 30542153
- PMCID: PMC6436733
- DOI: 10.1038/s41586-018-0781-z
Trapping biosynthetic acyl-enzyme intermediates with encoded 2,3-diaminopropionic acid
Abstract
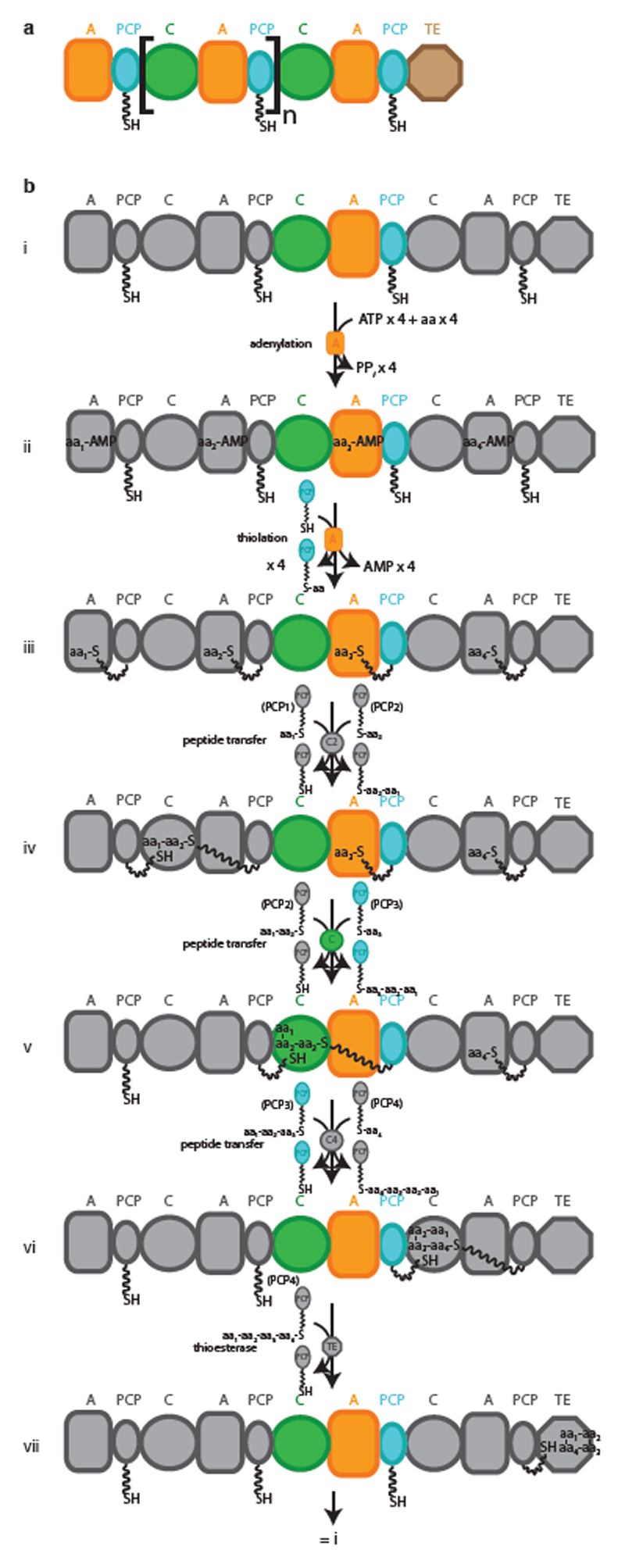
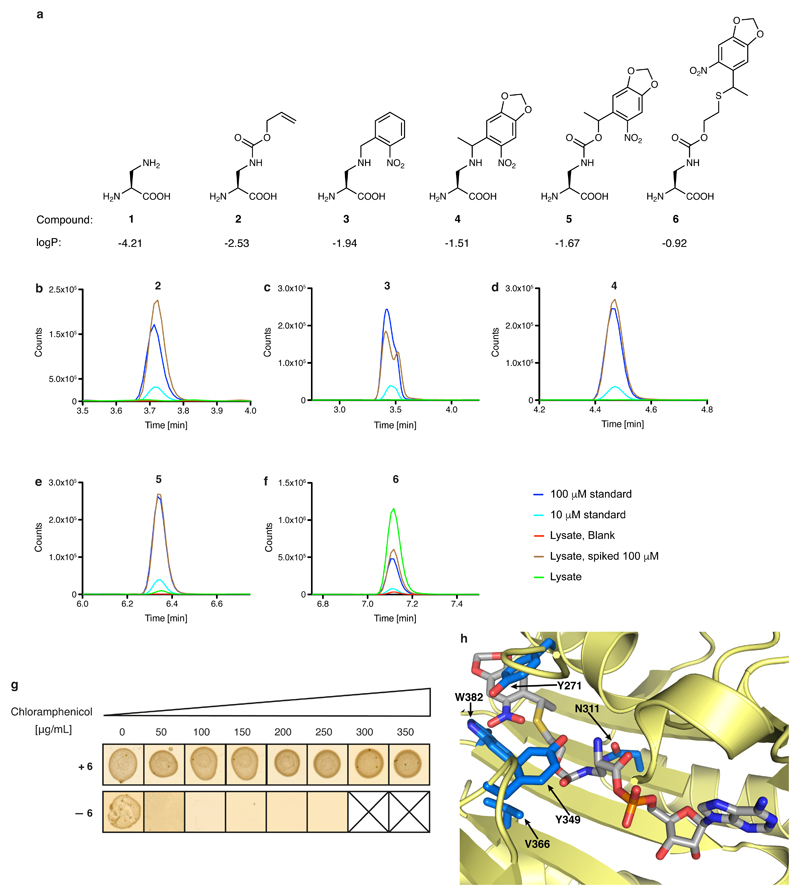
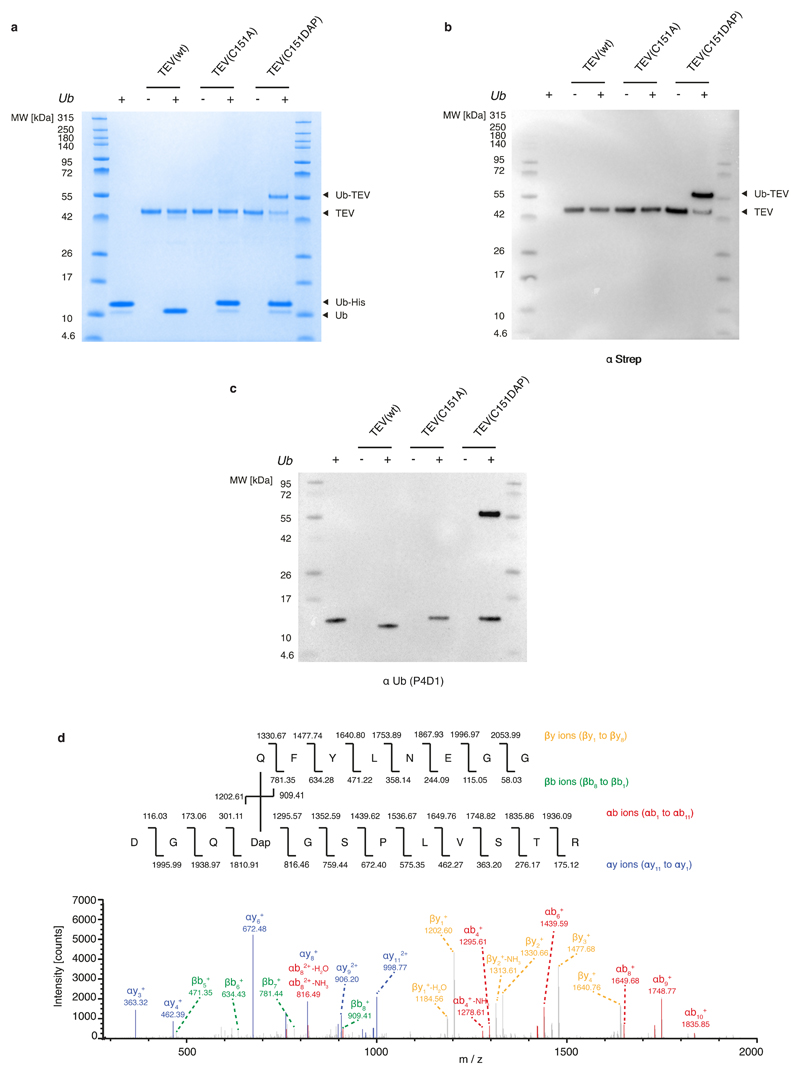
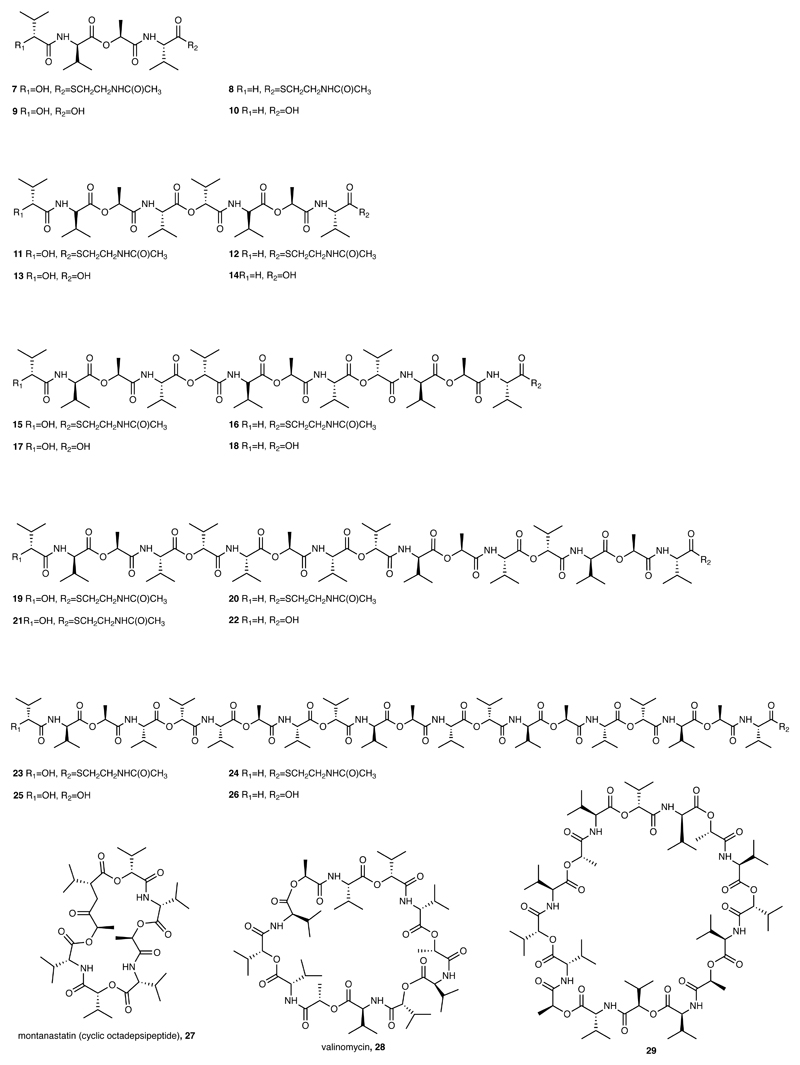
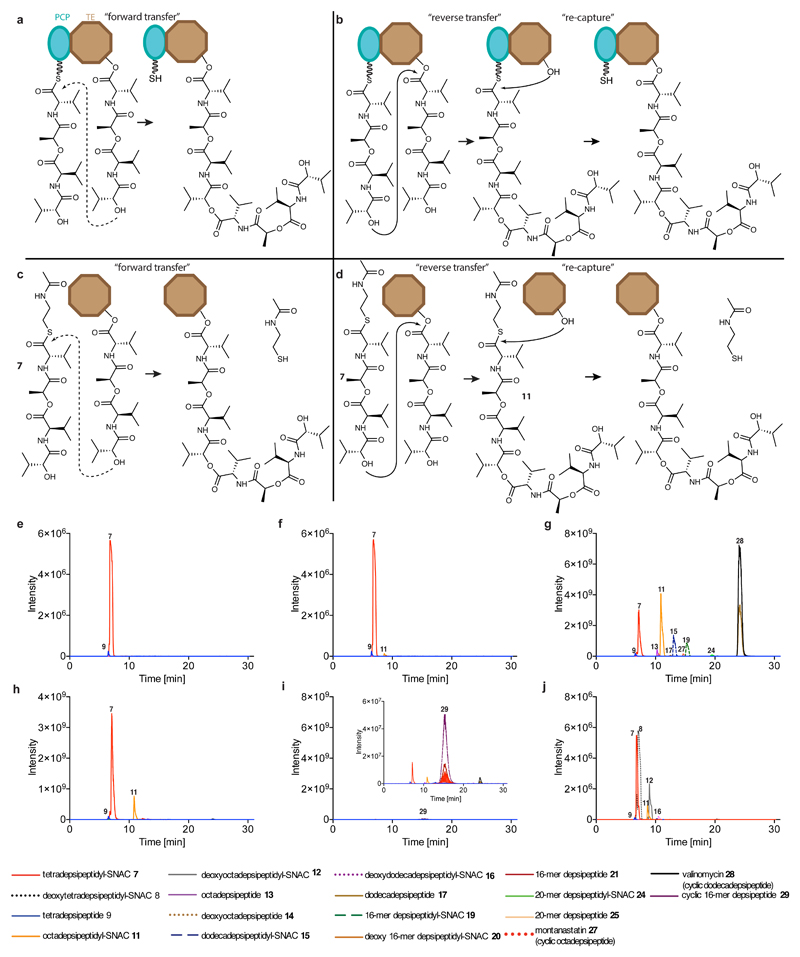
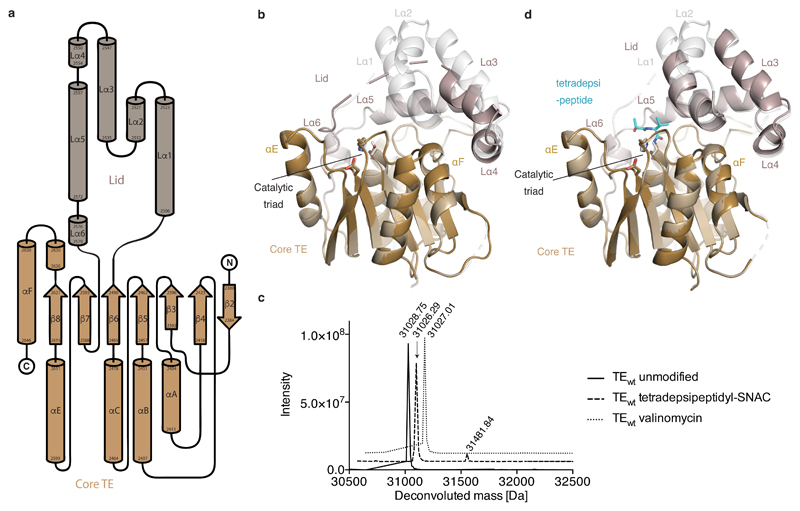
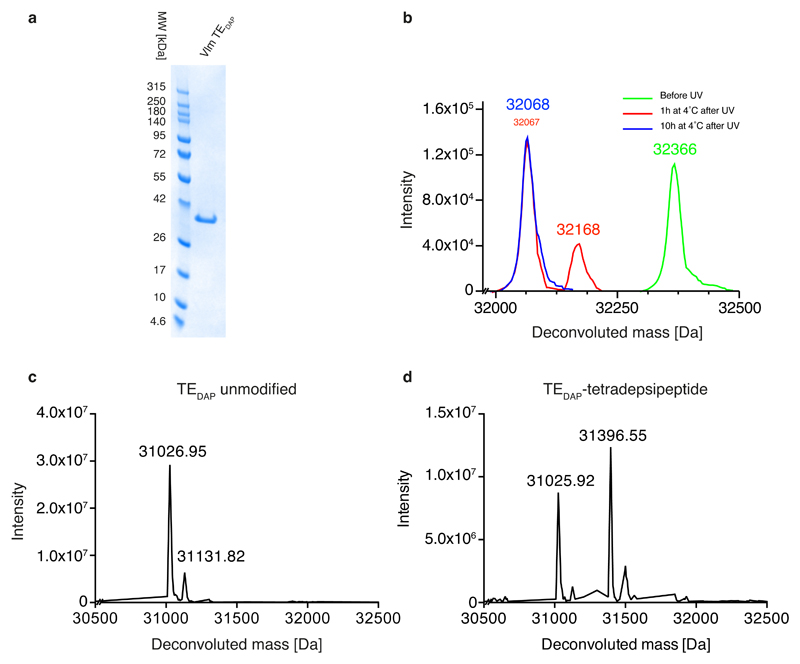
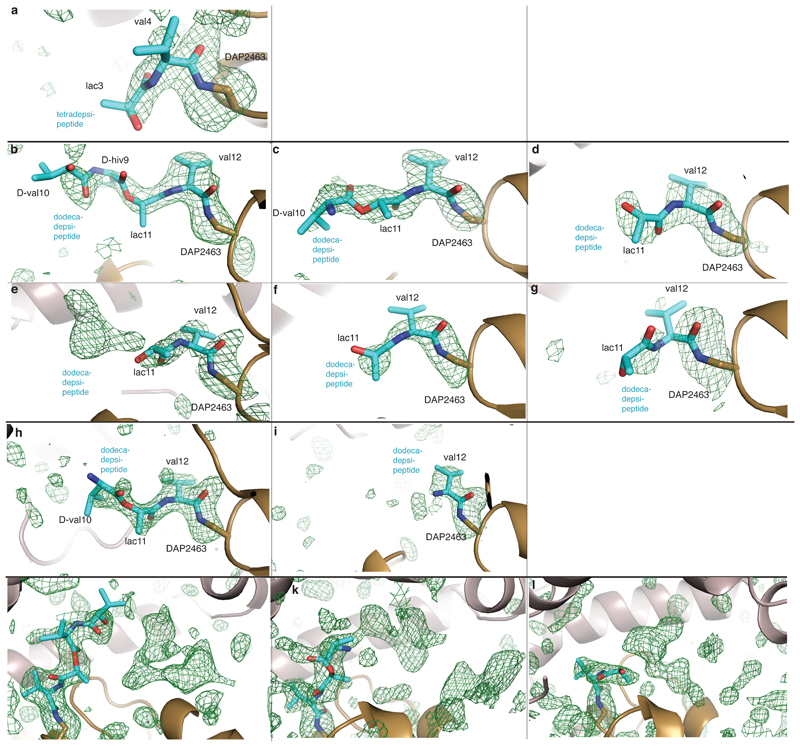
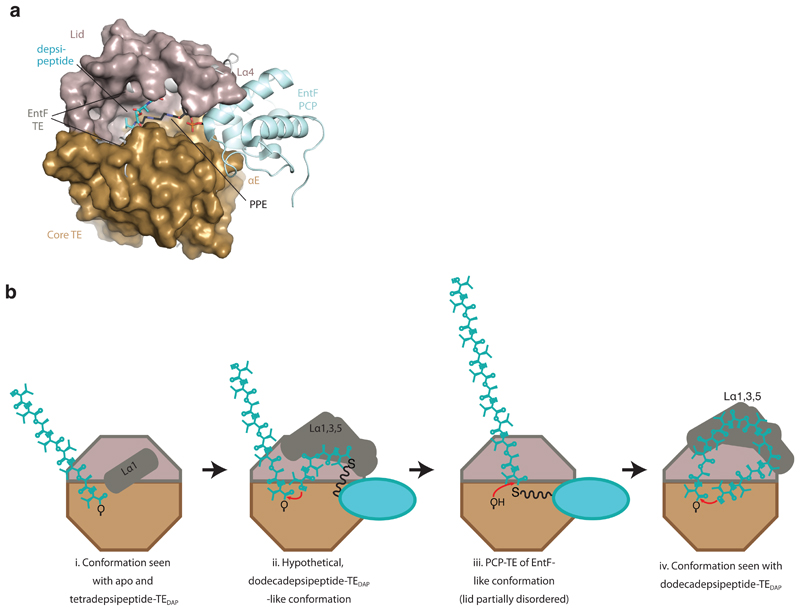
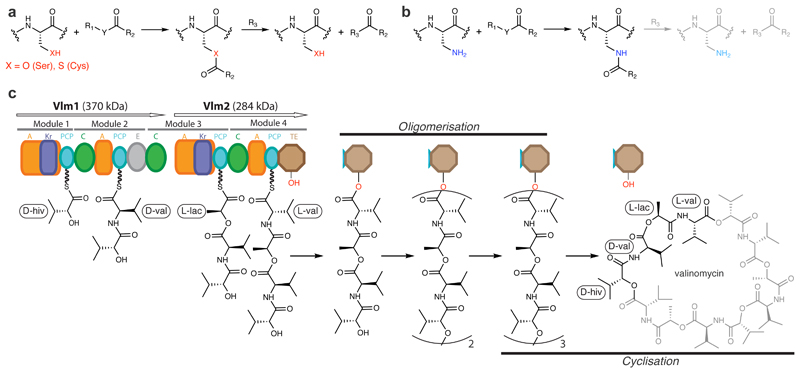
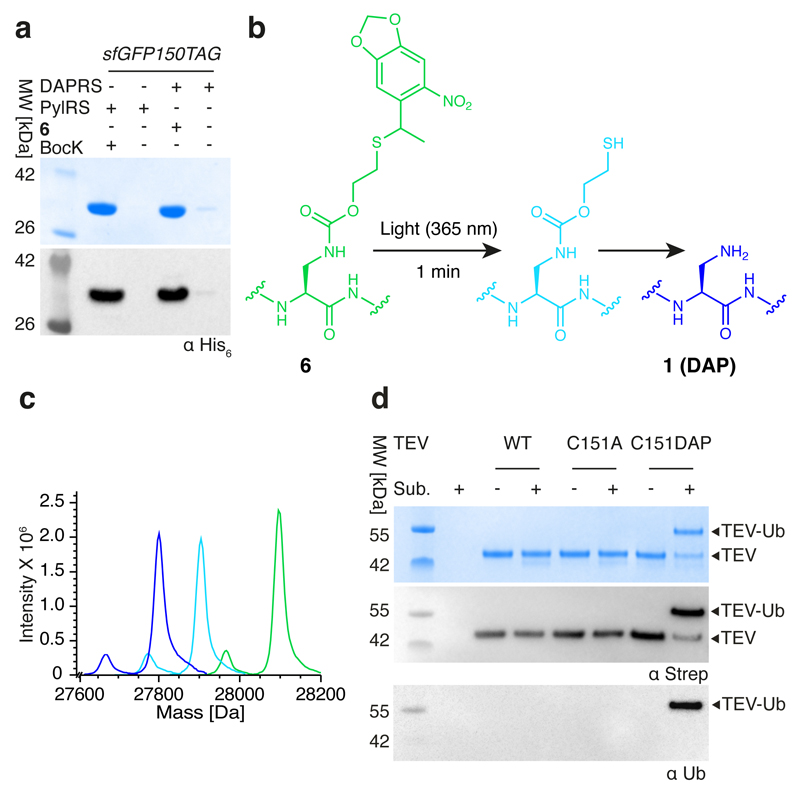
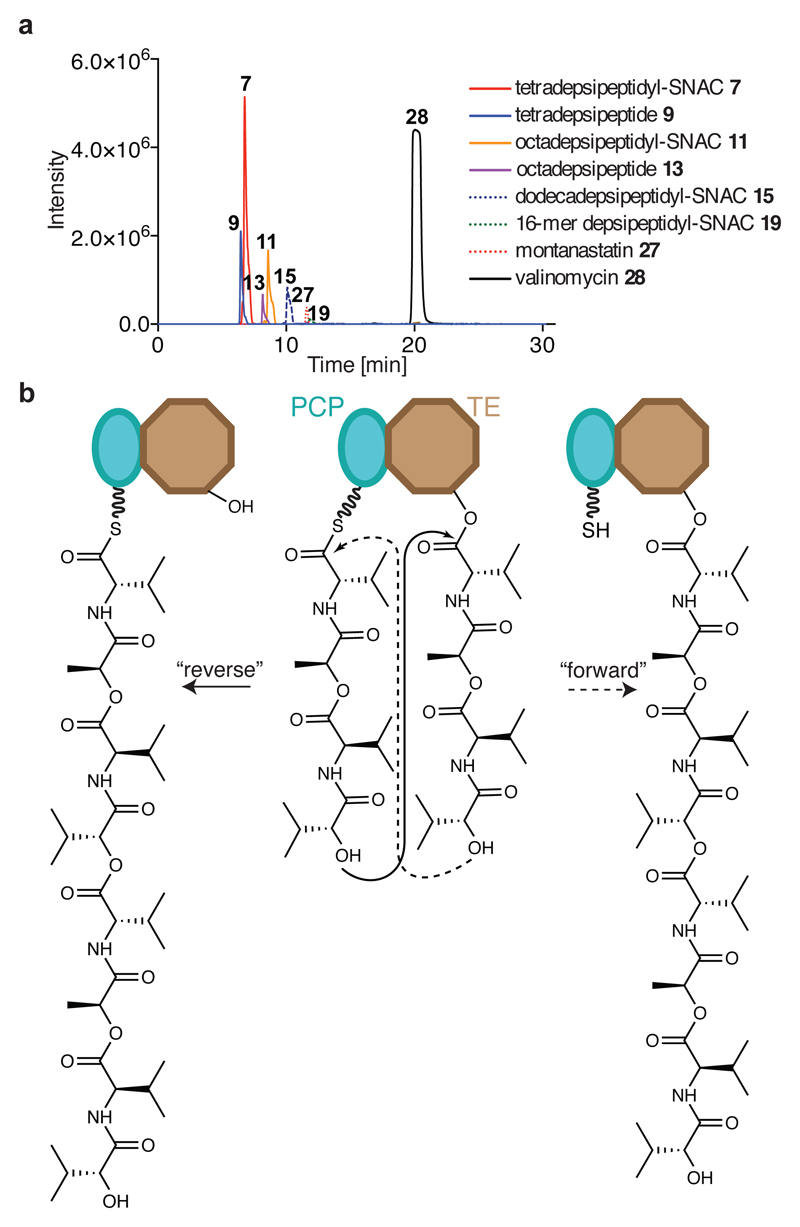
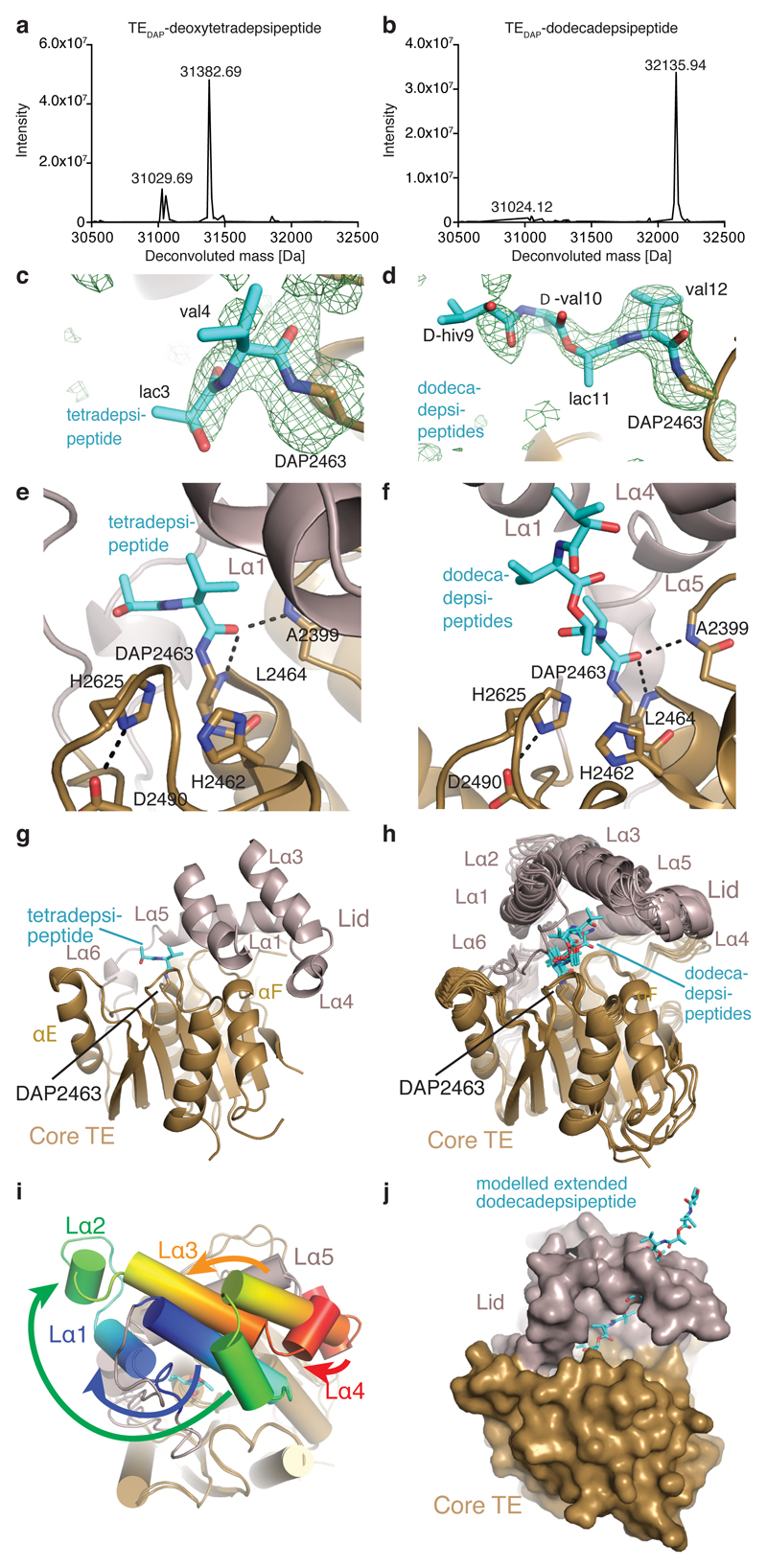
Many enzymes catalyse reactions that proceed through covalent acyl-enzyme (ester or thioester) intermediates1. These enzymes include serine hydrolases2,3 (encoded by one per cent of human genes, and including serine proteases and thioesterases), cysteine proteases (including caspases), and many components of the ubiquitination machinery4,5. Their important acyl-enzyme intermediates are unstable, commonly having half-lives of minutes to hours6. In some cases, acyl-enzyme complexes can be stabilized using substrate analogues or active-site mutations but, although these approaches can provide valuable insight7-10, they often result in complexes that are substantially non-native. Here we develop a strategy for incorporating 2,3-diaminopropionic acid (DAP) into recombinant proteins, via expansion of the genetic code11. We show that replacing catalytic cysteine or serine residues of enzymes with DAP permits their first-step reaction with native substrates, allowing the efficient capture of acyl-enzyme complexes that are linked through a stable amide bond. For one of these enzymes, the thioesterase domain of valinomycin synthetase12, we elucidate the biosynthetic pathway by which it progressively oligomerizes tetradepsipeptidyl substrates to a dodecadepsipeptidyl intermediate, which it then cyclizes to produce valinomycin. By trapping the first and last acyl-thioesterase intermediates in the catalytic cycle as DAP conjugates, we provide structural insight into how conformational changes in thioesterase domains of such nonribosomal peptide synthetases control the oligomerization and cyclization of linear substrates. The encoding of DAP will facilitate the characterization of diverse acyl-enzyme complexes, and may be extended to capturing the native substrates of transiently acylated proteins of unknown function.
Conflict of interest statement
Figures
Comment in
-
Enzymes engineered to trap reaction intermediates.Nature. 2019 Jan;565(7737):28-29. doi: 10.1038/d41586-018-07569-6. Nature. 2019. PMID: 30573786 No abstract available.
-
A trap for intermediate enzyme complexes.Nat Methods. 2019 Mar;16(3):217. doi: 10.1038/s41592-019-0339-5. Nat Methods. 2019. PMID: 30814698 No abstract available.
References
-
- Holliday GL, Mitchell JBO, Thornton JM. Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol. 2009;390:560–577. - PubMed
-
- Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. - PubMed
-
- Otto HH, Schirmeister T. Cysteine proteases and their inhibitors. Chem Rev. 1997;97:133–172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
