

Japanese Encephalitis Virus NS1' Protein Antagonizes Interferon Beta Production
- PMID: 30542978
- PMCID: PMC6335221
- DOI: 10.1007/s12250-018-0067-5
Japanese Encephalitis Virus NS1' Protein Antagonizes Interferon Beta Production
Abstract
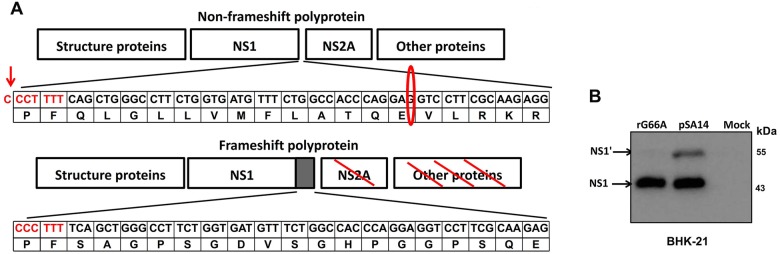
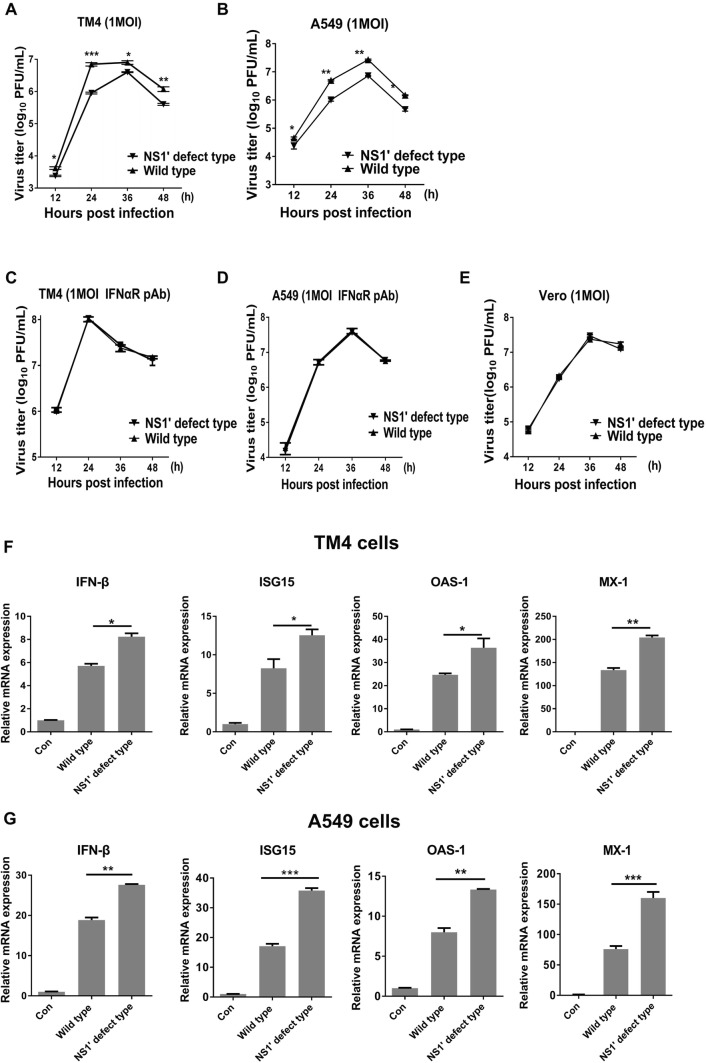
Japanese encephalitis virus (JEV) is a mosquito-borne virus and the major cause of viral encephalitis in Asia. NS1', a 52-amino acid C-terminal extension of NS1, is generated with a -1 programmed ribosomal frameshift and is only present in members of the Japanese encephalitis serogroup of flaviviruses. Previous studies demonstrated that NS1' plays a vital role in virulence, but the mechanism is unclear. In this study, an NS1' defected (rG66A) virus was generated. We found that rG66A virus was less virulent than its parent virus (pSA14) in wild-type mice. However, similar mortality caused by the two viruses was observed in an IFNAR knockout mouse model. Moreover, we found that rG66A virus induced a greater type I interferon (IFN) response than that by pSA14, and JEV NS1' significantly inhibited the production of IFN-β and IFN-stimulated genes. Taken together, our results reveal that NS1' plays a vital role in blocking type I IFN production to help JEV evade antiviral immunity and benefit viral replication.
Keywords: Immune evasion; Japanese encephalitis virus (JEV); NS1′; Type I interferon (IFN-I).
Conflict of interest statement
Conflict of interest
The authors declare no conflict of interests. All authors read and approved the final manuscript.
Animal and Human Rights Statement
Animal experiments in this study were approved by the Scientific Ethics Committee of Huazhong Agricultural University (permit number HZAUMO-2017-016).
Figures


Similar articles
-
Japanese Encephalitis Virus NS1' Protein Interacts with Host CDK1 Protein to Regulate Antiviral Response.Microbiol Spectr. 2021 Dec 22;9(3):e0166121. doi: 10.1128/Spectrum.01661-21. Epub 2021 Nov 10. Microbiol Spectr. 2021. PMID: 34756071 Free PMC article.
-
The Japanese Encephalitis Virus NS1' Protein Inhibits Type I IFN Production by Targeting MAVS.J Immunol. 2020 Mar 1;204(5):1287-1298. doi: 10.4049/jimmunol.1900946. Epub 2020 Jan 29. J Immunol. 2020. PMID: 31996459
-
The A66G back mutation in NS2A of JEV SA14-14-2 strain contributes to production of NS1' protein and the secreted NS1' can be used for diagnostic biomarker for virulent virus infection.Infect Genet Evol. 2015 Dec;36:116-125. doi: 10.1016/j.meegid.2015.09.013. Epub 2015 Sep 16. Infect Genet Evol. 2015. PMID: 26384477
-
The Japanese encephalitis virus NS1' protein facilitates virus infection in mosquitoes.PLoS Negl Trop Dis. 2025 Jan 27;19(1):e0012823. doi: 10.1371/journal.pntd.0012823. eCollection 2025 Jan. PLoS Negl Trop Dis. 2025. PMID: 39869646 Free PMC article.
-
Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor.Viruses. 2019 Jan 8;11(1):35. doi: 10.3390/v11010035. Viruses. 2019. PMID: 30625992 Free PMC article. Review.
Cited by
-
Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection.Viruses. 2022 Jun 20;14(6):1338. doi: 10.3390/v14061338. Viruses. 2022. PMID: 35746809 Free PMC article. Review.
-
Mechanisms Underlying Host Range Variation in Flavivirus: From Empirical Knowledge to Predictive Models.J Mol Evol. 2021 Jul;89(6):329-340. doi: 10.1007/s00239-021-10013-5. Epub 2021 May 31. J Mol Evol. 2021. PMID: 34059925 Review.
-
Japanese encephalitis virus inhibits superinfection of Zika virus in cells by the NS2B protein.J Virol. 2024 Mar 19;98(3):e0185923. doi: 10.1128/jvi.01859-23. Epub 2024 Feb 27. J Virol. 2024. PMID: 38411948 Free PMC article.
-
Efficient control of Japanese encephalitis virus in the central nervous system of infected pigs occurs in the absence of a pronounced inflammatory immune response.J Neuroinflammation. 2020 Oct 23;17(1):315. doi: 10.1186/s12974-020-01974-3. J Neuroinflammation. 2020. PMID: 33097065 Free PMC article.
-
Attenuating Mutations in Usutu Virus: Towards Understanding Orthoflavivirus Virulence Determinants and Live Attenuated Vaccine Design.Vaccines (Basel). 2025 May 3;13(5):495. doi: 10.3390/vaccines13050495. Vaccines (Basel). 2025. PMID: 40432107 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
