

Macrophage Mineralocorticoid Receptor Is a Pleiotropic Modulator of Myocardial Infarct Healing
- PMID: 30543467
- PMCID: PMC6291261
- DOI: 10.1161/HYPERTENSIONAHA.118.12162
Macrophage Mineralocorticoid Receptor Is a Pleiotropic Modulator of Myocardial Infarct Healing
Abstract
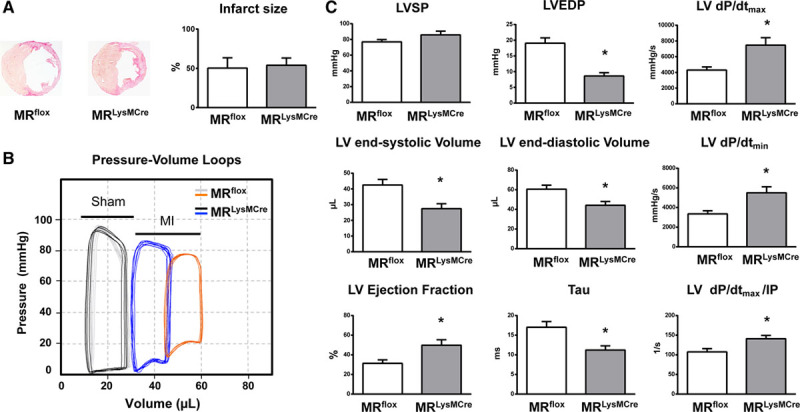
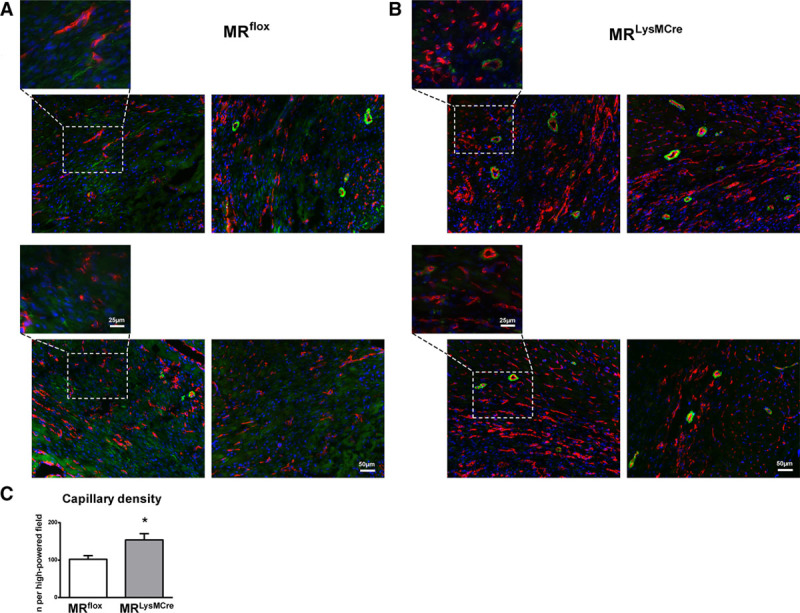
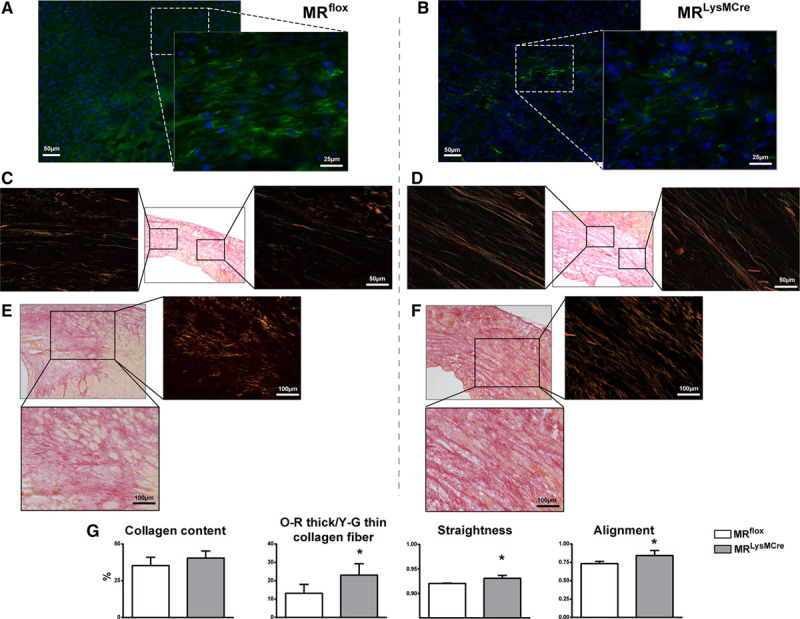
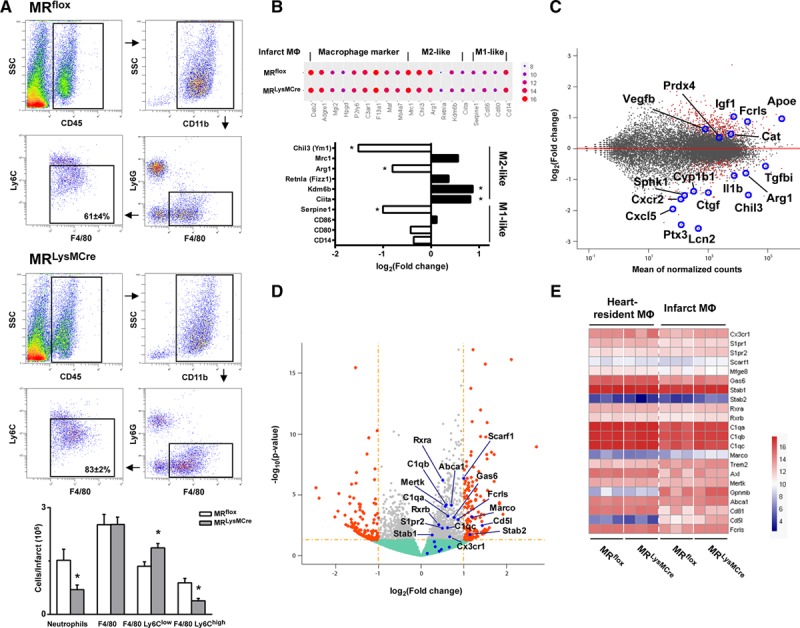
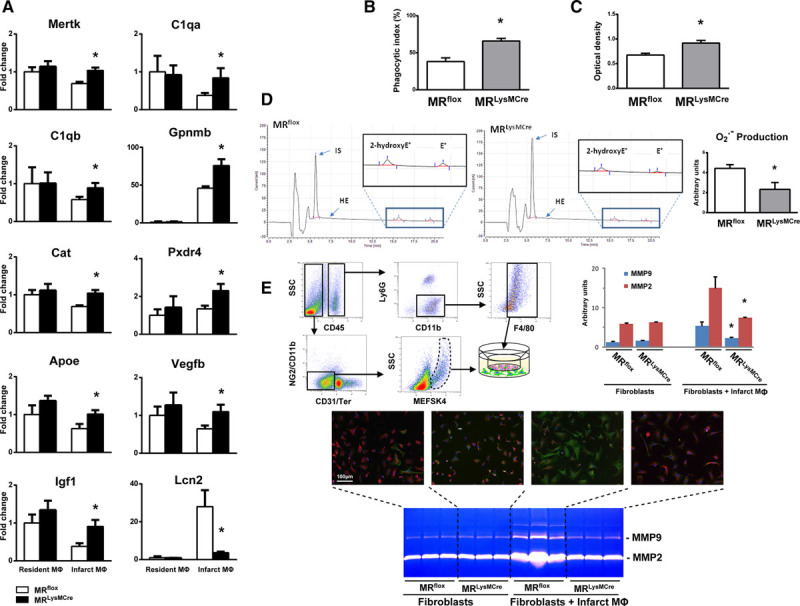
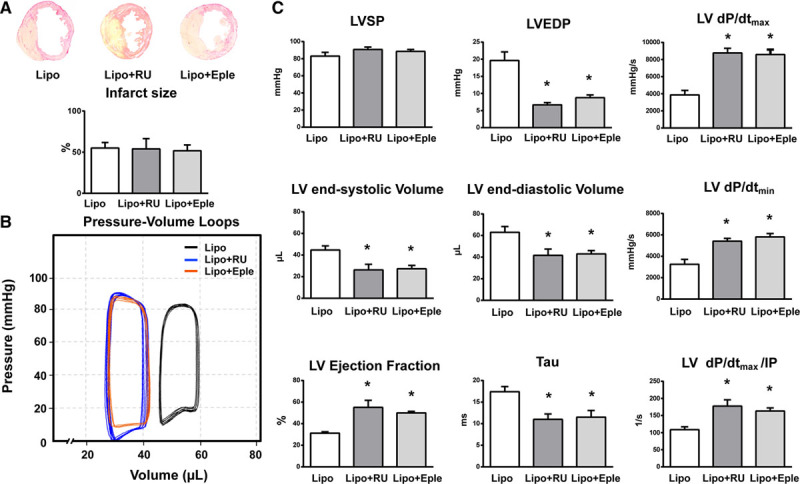
Myocardial infarction (MI) is a major cause of death worldwide. Here, we identify the macrophage MR (mineralocorticoid receptor) as a crucial pathogenic player in cardiac wound repair after MI. Seven days after left coronary artery ligation, mice with myeloid cell-restricted MR deficiency compared with WT (wild type) controls displayed improved cardiac function and remodeling associated with enhanced infarct neovascularization and scar maturation. Gene expression profiling of heart-resident and infarct macrophages revealed that MR deletion drives macrophage differentiation in the ischemic microenvironment toward a phenotype outside the M1/M2 paradigm, with regulation of multiple interrelated factors controlling wound healing and tissue repair. Mechanistic and functional data suggest that inactivation of the macrophage MR promotes myocardial infarct healing through enhanced efferocytosis of neutrophils, the suppression of free radical formation, and the modulation of fibroblast activation state. Crucially, targeted delivery of MR antagonists to macrophages, with a single administration of RU28318 or eplerenone-containing liposomes at the onset of MI, improved the healing response and protected against cardiac remodeling and functional deterioration, offering an effective and unique therapeutic strategy for cardiac repair.
Keywords: liposomes; macrophages; myocardial infarction; receptors, mineralocorticoid; wound healing.
Figures
Similar articles
-
Cardiac macrophages and apoptosis after myocardial infarction: effects of central MR blockade.Am J Physiol Regul Integr Comp Physiol. 2014 Oct 1;307(7):R879-87. doi: 10.1152/ajpregu.00075.2014. Epub 2014 Aug 6. Am J Physiol Regul Integr Comp Physiol. 2014. PMID: 25100076
-
Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response.Hypertension. 2008 Apr;51(4):905-14. doi: 10.1161/HYPERTENSIONAHA.107.100941. Epub 2008 Feb 25. Hypertension. 2008. PMID: 18299485
-
Lgr4 Governs a Pro-Inflammatory Program in Macrophages to Antagonize Post-Infarction Cardiac Repair.Circ Res. 2020 Sep 25;127(8):953-973. doi: 10.1161/CIRCRESAHA.119.315807. Epub 2020 Jun 30. Circ Res. 2020. PMID: 32600176
-
Aldosterone target organ protection by eplerenone.Mol Cell Endocrinol. 2004 Mar 31;217(1-2):229-38. doi: 10.1016/j.mce.2003.10.047. Mol Cell Endocrinol. 2004. PMID: 15134822 Review.
-
The cardioprotective effects of mineralocorticoid receptor antagonists.Pharmacol Ther. 2014 Apr;142(1):72-87. doi: 10.1016/j.pharmthera.2013.11.006. Epub 2013 Nov 23. Pharmacol Ther. 2014. PMID: 24275323 Review.
Cited by
-
New Perspectives on Sex Steroid and Mineralocorticoid Receptor Signaling in Cardiac Ischemic Injury.Front Physiol. 2022 Jun 29;13:896425. doi: 10.3389/fphys.2022.896425. eCollection 2022. Front Physiol. 2022. PMID: 35846011 Free PMC article. Review.
-
Mineralocorticoid receptor activation and antagonism in cardiovascular disease: cellular and molecular mechanisms.Kidney Int Suppl (2011). 2022 Apr;12(1):19-26. doi: 10.1016/j.kisu.2021.11.001. Epub 2022 Mar 18. Kidney Int Suppl (2011). 2022. PMID: 35529088 Free PMC article. Review.
-
Small animal models of heart failure.Cardiovasc Res. 2019 Nov 1;115(13):1838-1849. doi: 10.1093/cvr/cvz161. Cardiovasc Res. 2019. PMID: 31243437 Free PMC article. Review.
-
Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies.Eur Heart J. 2022 Jul 14;43(27):2549-2561. doi: 10.1093/eurheartj/ehac223. Eur Heart J. 2022. PMID: 35511857 Free PMC article. Review.
-
The role of endogenous Smad7 in regulating macrophage phenotype following myocardial infarction.FASEB J. 2022 Jul;36(7):e22400. doi: 10.1096/fj.202101956RR. FASEB J. 2022. PMID: 35695814 Free PMC article.
References
-
- Fraccarollo D, Galuppo P, Bauersachs J. Novel therapeutic approaches to post-infarction remodelling. Cardiovasc Res. 2012;94:293–303. doi: 10.1093/cvr/cvs109. - PubMed
-
- Pfeffer MA, Rutherford JD. Therapeutic attenuation of cardiac remodeling after acute myocardial infarction: a conversation with Marc A. Pfeffer, MD, PhD. Circulation. 2018;137:2430–2434. doi: 10.1161/CIRCULATIONAHA.118.033665. - PubMed
-
- Beygui F, Van Belle E, Ecollan P, Machecourt J, Hamm CW, Lopez De Sa E, Flather M, Verheugt FWA, Vicaut E, Zannad F, Pitt B, Montalescot G. Individual participant data analysis of two trials on aldosterone blockade in myocardial infarction. Heart. 2018;104:1843–1849. doi: 10.1136/heartjnl-2018–312950. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
