

Ribonuclease H1-targeted R-loops in surface antigen gene expression sites can direct trypanosome immune evasion
- PMID: 30543624
- PMCID: PMC6292569
- DOI: 10.1371/journal.pgen.1007729
Ribonuclease H1-targeted R-loops in surface antigen gene expression sites can direct trypanosome immune evasion
Abstract
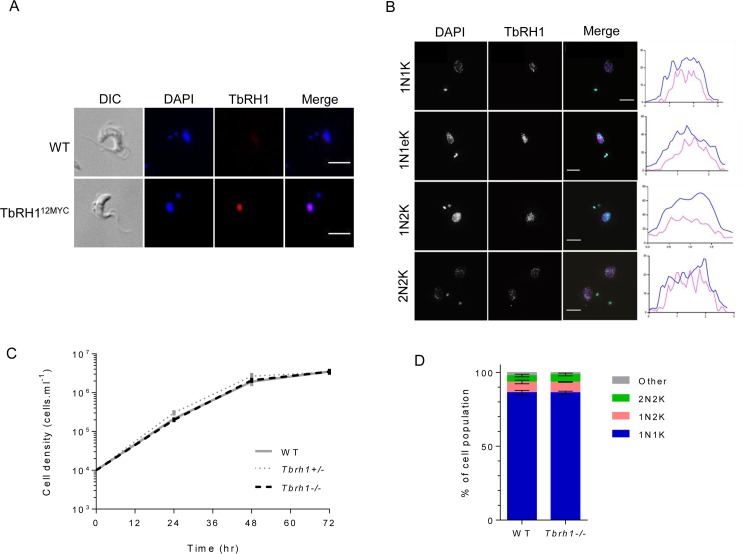
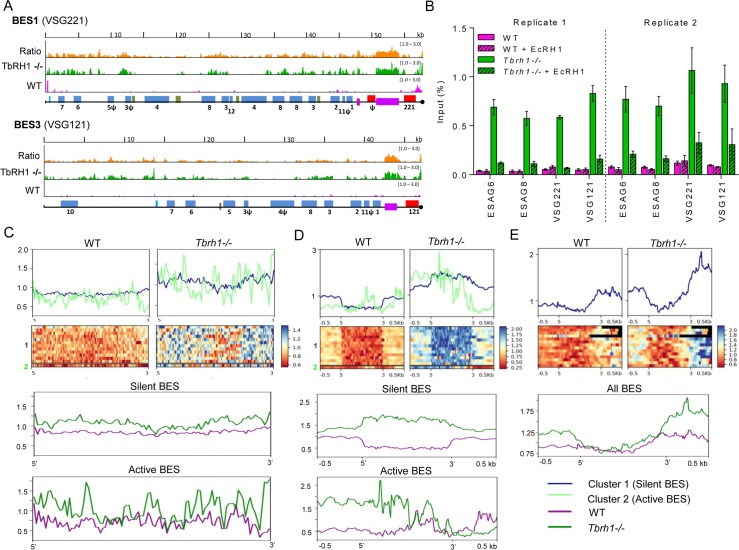
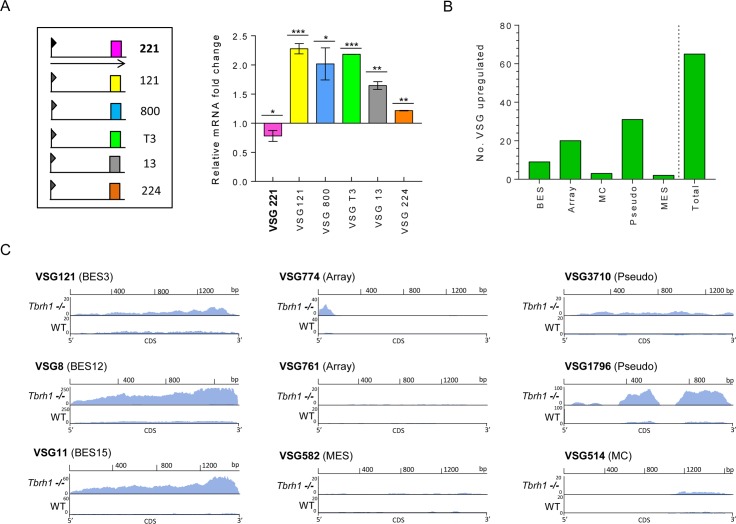
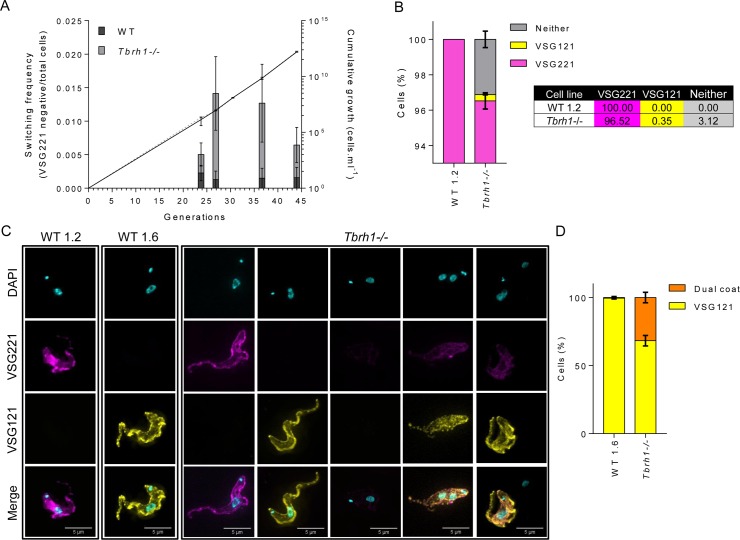
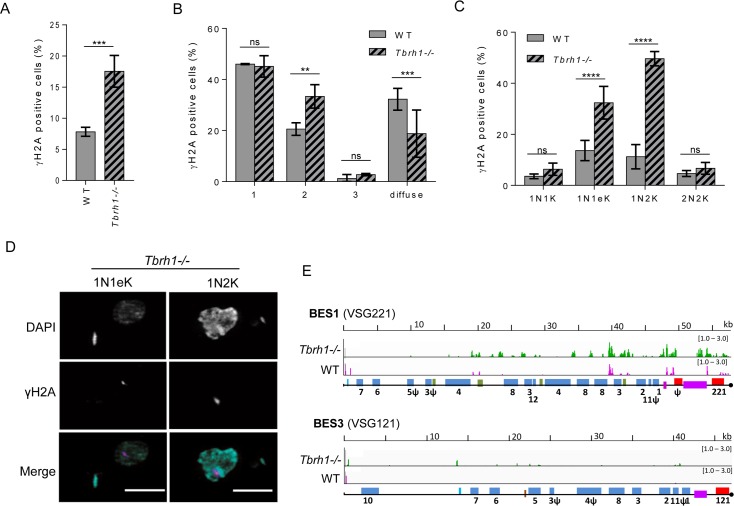
Switching of the Variant Surface Glycoprotein (VSG) in Trypanosoma brucei provides a crucial host immune evasion strategy that is catalysed both by transcription and recombination reactions, each operating within specialised telomeric VSG expression sites (ES). VSG switching is likely triggered by events focused on the single actively transcribed ES, from a repertoire of around 15, but the nature of such events is unclear. Here we show that RNA-DNA hybrids, called R-loops, form preferentially within sequences termed the 70 bp repeats in the actively transcribed ES, but spread throughout the active and inactive ES, in the absence of RNase H1, which degrades R-loops. Loss of RNase H1 also leads to increased levels of VSG coat switching and replication-associated genome damage, some of which accumulates within the active ES. This work indicates VSG ES architecture elicits R-loop formation, and that these RNA-DNA hybrids connect T. brucei immune evasion by transcription and recombination.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Comment in
-
R-loops modulate Trypanosome antigenic variation.PLoS Genet. 2018 Dec 13;14(12):e1007809. doi: 10.1371/journal.pgen.1007809. eCollection 2018 Dec. PLoS Genet. 2018. PMID: 30543632 Free PMC article. No abstract available.
Similar articles
-
Evaluation of mechanisms that may generate DNA lesions triggering antigenic variation in African trypanosomes.PLoS Pathog. 2018 Nov 15;14(11):e1007321. doi: 10.1371/journal.ppat.1007321. eCollection 2018 Nov. PLoS Pathog. 2018. PMID: 30440029 Free PMC article. Review.
-
Variant antigen diversity in Trypanosoma vivax is not driven by recombination.Nat Commun. 2020 Feb 12;11(1):844. doi: 10.1038/s41467-020-14575-8. Nat Commun. 2020. PMID: 32051413 Free PMC article.
-
Monoallelic expression and epigenetic inheritance sustained by a Trypanosoma brucei variant surface glycoprotein exclusion complex.Nat Commun. 2019 Jul 9;10(1):3023. doi: 10.1038/s41467-019-10823-8. Nat Commun. 2019. PMID: 31289266 Free PMC article.
-
Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species.Proc Natl Acad Sci U S A. 2012 Feb 28;109(9):3416-21. doi: 10.1073/pnas.1117313109. Epub 2012 Feb 13. Proc Natl Acad Sci U S A. 2012. PMID: 22331916 Free PMC article.
-
A Host-Pathogen Interaction Reduced to First Principles: Antigenic Variation in T. brucei.Results Probl Cell Differ. 2015;57:23-46. doi: 10.1007/978-3-319-20819-0_2. Results Probl Cell Differ. 2015. PMID: 26537376 Review.
Cited by
-
Trypanosoma brucei ATR Links DNA Damage Signaling during Antigenic Variation with Regulation of RNA Polymerase I-Transcribed Surface Antigens.Cell Rep. 2020 Jan 21;30(3):836-851.e5. doi: 10.1016/j.celrep.2019.12.049. Cell Rep. 2020. PMID: 31968257 Free PMC article.
-
Telomere and Subtelomere R-loops and Antigenic Variation in Trypanosomes.J Mol Biol. 2020 Jul 10;432(15):4167-4185. doi: 10.1016/j.jmb.2019.10.025. Epub 2019 Nov 2. J Mol Biol. 2020. PMID: 31682833 Free PMC article. Review.
-
Evaluation of mechanisms that may generate DNA lesions triggering antigenic variation in African trypanosomes.PLoS Pathog. 2018 Nov 15;14(11):e1007321. doi: 10.1371/journal.ppat.1007321. eCollection 2018 Nov. PLoS Pathog. 2018. PMID: 30440029 Free PMC article. Review.
-
Unraveling R-loops: The hidden drivers of inflammation and immune dysregulation.Medicine (Baltimore). 2025 Jun 13;104(24):e42833. doi: 10.1097/MD.0000000000042833. Medicine (Baltimore). 2025. PMID: 40527823 Free PMC article. Review.
-
R-loops modulate Trypanosome antigenic variation.PLoS Genet. 2018 Dec 13;14(12):e1007809. doi: 10.1371/journal.pgen.1007809. eCollection 2018 Dec. PLoS Genet. 2018. PMID: 30543632 Free PMC article. No abstract available.
References
-
- Yao MC, Chao JL, Cheng CY. Programmed Genome Rearrangements in Tetrahymena. Microbiology spectrum. 2014;2(6). 10.1128/microbiolspec.MDNA3-0012-2014 . - DOI - PubMed
-
- Sterkers Y, Crobu L, Lachaud L, Pages M, Bastien P. Parasexuality and mosaic aneuploidy in Leishmania: alternative genetics. Trends in parasitology. 2014;30(9):429–35. 10.1016/j.pt.2014.07.002 . - DOI - PubMed
-
- Ubeda JM, Raymond F, Mukherjee A, Plourde M, Gingras H, Roy G, et al. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS biology. 2014;12(5):e1001868 10.1371/journal.pbio.1001868 ; PubMed Central PMCID: PMC4028189. - DOI - PMC - PubMed
-
- Lee CS, Haber JE. Mating-type Gene Switching in Saccharomyces cerevisiae. Microbiology spectrum. 2015;3(2):MDNA3-0013-2014. 10.1128/microbiolspec.MDNA3-0013-2014 . - DOI - PubMed
-
- Klar AJ, Ishikawa K, Moore S. A Unique DNA Recombination Mechanism of the Mating/Cell-type Switching of Fission Yeasts: a Review. Microbiology spectrum. 2014;2(5). 10.1128/microbiolspec.MDNA3-0003-2014 . - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 104111/WT_/Wellcome Trust/United Kingdom
- BB/N016165/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M028909/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/K006495/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
